
Abstract
Traditionally, mitochondria are considered sites of energy production. However, recent studies have suggested that mitochondria are signaling organelles that are involved in intracellular interactions with other organelles. Remarkably, stressed mitochondria appear to induce a beneficial response that restores mitochondrial function and cellular homeostasis. These mitochondrial stress-centered signaling pathways have been rapidly elucidated in multiple organisms. In this review, we examine current perspectives on how mitochondria communicate with the rest of the cell, highlighting mitochondria-to-nucleus (mitonuclear) communication under various stresses. Our understanding of mitochondria as signaling organelles may provide new insights into disease susceptibility and lifespan extension.
Similar content being viewed by others

Introduction
‘They live inside me?’
Star Wars (1999)1
In the Star Wars universe, all cells contain intelligent life forms1,2 called midi-chlorians2. Life cannot exist without mid-chlorians, which form symbiotic relationships with host cells. The force speaks through the midi-chlorians, which connect the Living Force with the Cosmic Force3. Because the two are connected, they can communicate and bring peace to the galaxy.
Mitochondria, which inspired George Lucas, the director of the Star Wars, to invent midi-chlorians, are the basis of our lives. Although we have traditionally viewed mitochondria only as bioenergetic factories, their key function might be closely linked to their communication with the rest of the cell, similar to midi-chlorians. Recently, the molecular nature of multiple mitochondria-centric signals has begun to emerge. How and why mitochondria participate in signaling are still outstanding questions.
Understanding how mitochondria evolve through endosymbiosis provides important clues for understanding the genesis of signaling features in mitochondria. We will begin this review by briefly describing two fundamentally different models of mitochondrial evolution. We will not attempt to argue for either. Instead, we will focus on specific aspects of mitochondrial communication with the other genome-bearing organelle, the nucleus. We will focus on mitochondrial signals that are released or triggered by mitochondria and subsequently induce an adaptive response in the nucleus. From this perspective, we discuss how stressed mitochondria can communicate with the nucleus by generating a signal that modulates overall stress defenses, thereby promoting longevity and facilitating organismal adaptation to stress.
Mitochondria as communicating organelles
This is an extremely exciting time in mitochondrial biology. In the last two decades, some of the long-held secrets about mitochondria have begun to be elucidated. High-resolution imaging techniques allow both qualitative and quantitative studies of mitochondrial dynamics in live cells4. Unlike the static structures depicted in textbooks, we now view mitochondria as highly dynamic organelles that undergo constant fusion and fission events for optimal functioning5,6. In fact, mitochondria are uniquely active organelles that control self-repair and self-renewal for quality control in response to a wide range of stresses7. These extensive studies now portray mitochondria as multifaceted signaling organelles8. Remarkably, this viewpoint attempts to point out new aspects of mitochondrial stress signaling and addresses several open questions: (1) How do mitochondria act when they are in trouble? (2) How do mitochondria communicate stress to the nucleus? (3) How can the mitochondrial stress response manage longevity and regulate disease susceptibility?
Mitochondria are mysterious organelles that evolved from endosymbionts
The origin of mitochondria is a hard evolutionary problem, intimately intertwined with the origin of eukaryotes, known as eukaryogenesis9,10 Currently, there are two major evolutionary theories regarding the origin of mitochondria: the ‘mitochondria-early’ and ‘mitochondria-late’ scenarios9 (Fig. 1). In the ‘mitochondria-early’ scenario, mitochondrial acquisition was the initial trigger for eukaryogenesis11. In contrast, in the ‘mitochondria-late’ scenario, mitochondrial symbiosis was the final step in eukaryogenesis12.

The evolutionary models for the endosymbiotic origin of mitochondria are generally classified as ‘mitochondria-early’ and ‘mitochondria-late’ scenarios depending on the timing of mitochondrial endosymbiosis during the process of eukaryogenesis. The ‘mitochondria-early’ model assumes metabolic symbiosis between prokaryotic archaea and α-proteobacteria. In this scenario, mitochondria were the first complex feature acquired within eukaryotic cells. In contrast, the ‘mitochondria-late’ hypothesis purports amitochondriate eukaryotes as the host engulfing mitochondrial ancestor. In this scenario, hosts with some eukaryotic features, referred to as ‘proto-eukaryotes’, engulf bacterial endosymbionts that become mitochondria. Both models include extensive gene transfer from the α-proteobacterium to the host and the coevolution of organellar and endosymbiont genomes in the host-endosymbiont complex. The figures were modified from ref. 9.
The hydrogen bond hypothesis proposed by Műller and Martin in 1998 led to widespread acceptance of the ‘mitochondria-early’ hypothesis12. In this view, the host was a hydrogen-consuming anaerobic archaeon, and the symbiont was a metabolically sophisticated α-proteobacterium capable of surviving in both aerobic and anaerobic conditions9. The products from the α-proteobacterium, such as hydrogen and carbon dioxide, were utilized by methane-producing archaea. Eventually, the archaeon enclosed the α-proteobacterium probably due to the strong selective force of host–endosymbiont energy dependence. This hypothesis is testable in principle. There is no need for evolutionary intervention; rather, this approach provides a selection-based explanation for why the host needs its endosymbiont.
However, the ‘mitochondria-late’ scenario places mitochondrial acquisition late in eukaryogenesis. This hypothesis posits that most significant features of eukaryotes could have arisen prior to the evolution of the mitochondrion13. According to this model, Archezoa were eukaryotes with no mitochondria14. This traditional theory is a favored model for explaining the origin of mitochondria, which is a defining event in the evolution of eukaryotes15. This theory seems to be a reasonable explanation, considering that the cytoskeleton in host cells can facilitate the uptake of bacterial endosymbionts9.
Regardless of how primordial mitochondria truly evolved, both models involve extensive gene transfer from the α-proteobacterium to the archaeal host. Although mitochondria contain their own circular genome, the majority of proteins in mitochondria are actually encoded by the nuclear genome. This gene loss may have allowed mitochondria to (1) sense and (2) respond to cellular and environmental stressors and (3) produce various signals that can tune the functions of other organelles, including the nucleus. Accordingly, this evolutionary tension to transfer gene to nucleus would rather lead mitochondria to control nucleus via mitonuclear communication.
Mitochondrial signals are transmitted to the nucleus during stress
For mutual benefit, mitochondria contribute to maintaining a symbiotic relationship with the rest of the cell. It is not surprising that mitochondria transmit retrograde signals, which in turn protect against their own dysfunction. These signals can arise from various mitochondrial compartments and reach multiple cellular components, including the nucleus and cytoplasm. Interestingly, they can even travel to distant cells via systemic circulation16.
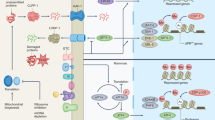
To date, only a small number of signaling molecules and signaling pathways have been investigated. One of the well-studied signaling cascades is cytochrome c (cyt c) release from mitochondria in multiple proapoptotic stimuli17. Presently, it is believed that the release of cyt c from mitochondria into the cytosol is a critical event in the apoptotic pathway of cell death18,19. Recent evidence suggests however that cyt c release can be part of defense mechanism against stresses20,21. Besides this activation of cytosolic signaling pathways, there are tantalizing clues that stressed mitochondria generate retrograde signals to the nucleus. Remarkably, rather than being harmful, emitted signals from stressed mitochondria appear to induce a wide-ranging protective cellular state by regulating nuclear gene expression (Fig. 2a).

a Stressed mitochondria can transport signaling molecules to the nucleus, where they either directly or indirectly regulate the expression of nuclear genes to trigger an adaptive stress response. b In the fed state, mitochondrial acetyl-CoA is delivered into the nucleus and utilized for histone acetylation in the nucleus for cell growth and proliferation. Stresses, including electron transport chain (ETC) dysfunction and nutrient stress, reduce the level of nucleocytosolic acetyl-CoA and limit histone acetylation. Such alterations in acetyl-CoA availability subsequently result in the induction of autophagy and promote survival under stress. c Mitochondrial stress generates reactive oxygen species (ROS) within the mitochondria. Mitochondrial ROS can trigger transcription-activating signaling cascades through redox-dependent modifications of proteins either directly or indirectly by activating ROS sensors. These ROS-activating signaling pathways lead to the expression of genes that mediate the adaptive response in the nucleus. d Calcium ions (Ca2+) can be released from mitochondria in response to many mitochondrial stresses, including depolarization of the mitochondrial membrane potential. Released Ca2+ triggers many different cellular pathways to stimulate transcription factors through Ca2+-sensitive protein kinases, phosphatases, and transcriptional repressors. Ca2+ signaling controls gene transcription to improve calcium homeostasis and mitochondrial metabolism. e The mitochondrial unfolded protein response (UPRmt) is a protective transcriptional response that enhances the expression of genes involved in the mitochondrial protein quality control system and stress defense. Various mitochondrial insults, including mitochondrial proteotaxic stresses and mitonuclear imbalance, which is an imbalance between the mitochondrial proteins encoded by the nucleus and those that are mitochondrially encoded, can trigger the UPRmt through a pleiotropic retrograde response to restore mitochondrial homeostasis. f The integrated stress response (ISR) is an adaptive stress pathway that is interconnected with various pathways under different stresses. Under diverse stress conditions, several kinases, including HRIs, are activated. These signals converge to phosphorylate the translation initiation factor eIF2α. The phosphorylation of eIF2α induces both general inhibition of protein translation and selective activation of a set of mRNAs that possess uORFs, such as ATF4. ATF4 in the nucleus binds to a transcription factor to activate genes involved in stress adaptation and mitochondrial function. eIF2α, α-subunit of eukaryotic initiation factor 2; ATF4, activating transcription factor 4.
Acetyl-coenzyme A
Metabolically, it is no surprise that metabolites play an essential role in signaling pathways and allow for mitochondria–nucleus communication. One possible messenger is acetyl-coenzyme A (acetyl-CoA), which is readily synthesized from acetate8. From archaea to mammals, acetyl-CoA occupies a central position in carbon metabolism; it exists at the intersection of catabolic and anabolic metabolism and is a key determinant of protein acetylation22.
Acetyl-CoA is predominantly produced in the mitochondria and can be transferred into the cytoplasm in the form of citrate (Fig. 2b). Accumulating evidence suggests that gene expression is determined by the concentration of acetyl-CoA or the ratio of acetyl-CoA to coenzyme CoA (acetyl-CoA/CoA) in the cellular compartment23,24. When nutrients are abundant, transcription is globally activated25, and the transcription of genes involved in cell growth and replication is preferentially promoted26. In contrast, when energy is limited, there is a greater need for acetyl-CoA as a fuel source in the mitochondria. Accordingly, the depletion of nucleocytosolic acetyl-CoA decreases histone acetylation and causes chromatin condensation. For survival, this nutrient stress activates autophagy genes27. Remarkably, a wide range of mitochondrial stresses, including electron transport chain (ETC) defects, have similar consequences at the organism level28.
These recent findings have rekindled interest in mitochondrial messengers that induce an adaptive response in the nucleus; these messengers were first discovered by Nobel laureates Jacob and Monod in the 1960s29. An intriguing possibility is that our cells have evolved to use acetyl-CoA as a messenger from mitochondria to the nucleus, which in turn determine cell fate through both metabolic and epigenetic mechanisms.
Reactive oxygen species
We cannot survive without oxygen. Unfortunately, oxygen is naturally dangerous to humans due to its electronic structure. When first discovered, reactive oxygen species (ROS)30 were onl considered dangerous reactive molecules. With intensive research over the past few decades, this view has been supplanted by the concept that ROS can function as messengers in numerous signaling pathways31. One of the clear examples came from studies in Caenorhabditis elegans (C. elegans). Interestingly, metabolic stress triggers a compensatory increase in mitochondrial respiration and increases ROS production, which ultimately promotes stress resistance and even extends the lifespan of cells32.
Why would stressed mitochondria promote stress resistance? At present, there are no definitive answers. One possibility is that in response to mild stress, mitochondria might release ROS as messenger molecules to trigger a cascade of cellular events that ultimately protect stressed cells through mitochondria-to-nucleus retrograde signaling33 (Fig. 2c). ROS may act as dose-dependent stress codes34 that are characterized by spatial and temporal patterns. There is a growing body of evidence suggesting that stressed mitochondria release ROS resulting in altered gene expression through a variety of signaling pathways35. In particular, redox-activated proteins appear to be involved in this ROS-mediated mitonuclear communication36. For example, posttranslational modification of redox-dependent thiols in regulatory proteins can affect the function of various transcription factors, including nuclear factor erythroid 2-related factor 2 (NFE2L2; also known as NRF2)36. This modification of master transcription factors subsequently upregulates the expression of genes involved in defense response and drives beneficial adaptive responses to mitochondrial stress.
Clearly, ROS are predominantly generated and accumulate in mitochondria. However, all proteins supporting antioxidant reactions are encoded in the nucleus, not in the mitochondria36. Eukaryotes may have evolved specific mechanisms that coordinate the expression of antioxidant gene sets by relying on retrograde signaling messengers from mitochondria to nucleus on demand37. These mechanisms are worth investigating. These discoveries could lead to the development of new strategies to elucidate the ROS stress code and facilitate therapeutic interventions (see below).
Calcium ions
In 1953, it was demonstrated that mitochondria could transport calcium ions (Ca2+)37. Since then, intense research has defined the role of mitochondria in the regulation of Ca2+ homeostasis. Mitochondrial Ca2+ signaling has emerged as an important player in tuning and optimizing the state of mitochondria in response to dynamic changes38. From this perspective, mitochondria act as Ca2+-dependent effectors involved in a wide range of cellular processes, including cell death and metabolism39.
Over the last few decades, the key role of mitochondrial Ca2+ in signaling has been elucidated by several groups. A vast range of mitochondrial stresses, such as the loss of mtDNA40, electron transport chain (ETC) deficiency34, and mitochondrial uncoupling40,41, can trigger the excessive release of Ca2+ from the mitochondria to the cytosol (Fig. 2d). Owing to the dynamic properties of Ca2+, its release from mitochondria can directly trigger multiple signaling pathways that transmit signals to the nucleus. For example, free Ca2+ in the cytosol can bind to multifunctional Ca2+-binding molecules, such as calmodulin (CaM)42,43. Once bound to Ca2+, CaM activates the calcium signal transduction pathway via the modification of its interactions with many target proteins, including protein kinases and protein phosphatases. These signals subsequently activate the transcription of specific genes, mainly through targeting transcription factors. Thus, Ca2+ in non-mitochondrial areas can affect the transcriptional outcome, which ultimately promotes calcium homeostasis and restores mitochondrial function in response to stress.
However, there has also been considerable interest in the harmful effects of calcium since the discovery of the calcium paradox in the 1960s44. Increased mitochondrial Ca2+ influx is a potent initiator of apoptosis45, necrosis46 and mitophagy47. As a vital secondary messenger, Ca2+ is a double-edged sword. A complete understanding of the pathways regulating mitochondrial Ca2+ homeostasis will be crucial for characterizing mitochondrial Ca2+ signaling and its contribution to the processes of aging and disease pathogenesis.
Mitochondrial unfolded protein response
Most mitochondrial proteins (~99%) are encoded within the nuclear genome, synthesized by cytosolic ribosomes, and imported into mitochondria as mitochondrial precursor proteins48. Once translocated to the mitochondria, precursor polypeptides are delivered to their destination, where they fold efficiently into their unique functional structure. Mitochondria actually possess a sophisticated protein quality control system that mainly comprises molecular chaperones and quality control proteases49. When the capacity of the protein quality control system reaches its limit, mitochondria become susceptible to the deleterious effects of protein aggregation and misfolding. Aberrant mitochondrial protein folding and aggregation have been linked to a plethora of human diseases including Parkinson’s disease50.
Responding to these stresses, mitochondria have evolved an organelle-specific defense system known as the mitochondrial unfolded protein response (UPRmt)51 (Fig. 2e). The UPRmt is a mitochondria-to-nucleus signaling pathway that is activated to restore mitochondrial function when misfolded proteins accumulate in the mitochondria51. A broad range of mitochondrial stresses, including imbalances in nuclear- and mitochondrial-encoded proteins52, can also initiate the UPRmt pathway (see ref. 53 and references therein). Indeed, the UPRmt is conserved in many organisms from worms to mammals51,54. In the UPRmt pathway, various components are activated for retrograde signaling from the mitochondria to the nucleus55. In particular, the UPRmt can activate transcription factors, such as C/EBP homologous protein (CHOP) and CCAAT enhancer binding protein beta (C/EBPβ), that in turn increase the expression of UPRmt genes. Activation of the UPRmt pathway enhances the adaptive stress response and helps maintain homeostasis by inducing the transcription of mitochondrial molecular chaperones and proteases.
Evidence suggests that UPRmt provides a specialized role in diverse processes governing mitochondrial homeostasis. For example, stressed mitochondria may be repaired through the UPRmt or eliminated by mitophagy56, depending upon the nature and severity of the mitochondrial dysfunction. Taken together, these observations suggested that UPRmt outputs can contribute increased stress resistance and longevity (see below). The mechanisms by which mitochondria utilize these stress responses are beginning to be understood, but the details remain unclear.
Integrated stress response
Considering energy economy, protein synthesis in eukaryotic cells is quite expensive. Consequently, this process needs to be performed at the right time, in the right place, and to the right extent. To maintain homeostasis, the translatome must be reprogrammed in response to diverse intercellular and environmental inputs. However, the exact cost and detailed molecular mechanism of protein synthesis in eukaryotes have not been clearly determined57.
Interestingly, mitochondrial dysfunction is associated with the activation of the integrated stress response (ISR), a signaling pathway that reshapes the translatome in response to stress43 (Fig. 2f). More generally, the ISR is a retrograde signaling pathway that integrates different stimuli from many organelles, including mitochondria, the endoplasmic reticulum (ER), and the cytoplasm, such that communication between organelles helps determine cell fate. The ISR combines a variety of stresses, including mitochondrial and intercellular perturbations58. Interestingly, the mitochondrial ISR has opposite effects. To resolve stress, the ISR inhibits general protein translation while favoring the translation of a small subset of mRNAs59. In the ISR, the α-subunit of eukaryotic initiation factor 2 (eIF2α) appears to be a central hub. The activation of eIF2α represses general translation via a cap-dependent mechanism. Intriguingly, certain proteins are still translated under stress conditions. Why only a limited set of genes resist translational repression by the ISR is an interesting question. Thus, it is intriguing to consider the mechanism underlying this paradoxical synthesis of stress response and how the ISR affects to maintain or reestablish physiological homeostasis.
Typically, translational control occurs primarily during initiation60. However, multiple genome-wide analyses, including those utilizing ribosomal footprinting approaches61, have demonstrated a novel mechanism of translational regulation by short, upstream open reading frames (uORFs) in the 5′ untranslated region (5’UTR)62. Recent evidence suggests that the induced genes during the ISR appear to contain multiple uORFs located 5′ to the coding sequence (CDS). One well-studied example involves activating transcription factor 4 (ATF4)63. In mammals, ATF4 messenger RNA (mRNA) has two uORFs before the initiation codon. In nonstressed cells, the 5′ proximal uORF (uORF1) facilitates ribosome scanning, which reinitiates at the downstream frame (uORF2) that blocks ATF4 expression. In stressed cells, high levels of eIF2α phosphorylation delay the capacitation of scanning ribosomes, inducing the inhibition of uORF2 and reinitiation at the ATF4 coding region64,65. These activated ATF4 subsequently induces the transcription of various stress response genes through activation of transcription factors, such as ATF566 and CHOP67.
Intriguingly, there is growing scientific interest in another view of this paradoxical outcome in the ISR. It is generally thought that the vast majority of eukaryotic mRNAs initiate translation through a cap-dependent manner. However, under stress conditions, the cap-independent translation driven by internal ribosome entry site (IRES) can serve as an alternative mechanism for protein production (see68 and references therein). It is possible that certain stresses shut down eukaryotic translation initiation factor 4E (eIF4E)-mediated translation in a cap-dependent manner while activate IRES-mediated translation by recruiting ribosomes for translation69. The full extent of these two opposing outcomes of ISR and their impact in stress resistance remains unexplored.
Over the past decade, genome-wide studies have clearly revealed the existence of widespread unannotated open reading frames (ORFs) throughout the genome. The pioneering work that stemmed from this discovery revealed a hidden proteome consisting of many categories of novel proteins from noncanonical ORFs70 (see below). There will be much to learn in this exciting new phase of research on the biology of translation.
Mitochondria-derived peptides as mitochondrial messengers
Emergence of small genes within ORFs
With all the excitement comes a need for caution. Approximately 20 years ago, the Human Genome Project (HGP), which includes an estimated 20,000 to 25,000 genes encoding functional proteins, was successfully completed71. While this vast project on decoding the bulk of human DNA has served us well in describing our genome, it also could constrain our understanding. Modern sequencing technologies have revealed additional complexity in the protein-coding capacity of our genome (see70 and references therein).
Recent breakthroughs in translatome proteomics have led to the explosive discovery of additional ORFs. Currently, 194,407 human-translated noncanonical ORFs have been predicted and experimentally verified70,72,73,74. Among the various types of noncanonical ORFs, small ORFs (sORFs) have been extensively highlighted. The protein products, known as sORF-encoded polypeptides (SEPs), are smaller than 100 amino acids in length. The search for hidden small proteins is invigorating the field of translatome proteomics. Therefore, it has prompted us to revisit our understanding of the eukaryotic gene model75,76.
Discovery of bioactive sORFs in the mitochondrial genome
Although SEPs in the nuclear genome are intriguing, their presence in the mitochondrial genome remains uncertain. One type of candidate SEP in the mitochondria is mitochondria-derived peptides (MDPs)77. MDPs are a class of SEPs. By now, eight MDPs have been identified from ribosomal RNA (rRNA) regions of mitochondrial genome, such as humanin78, mitochondrial open reading frame of 12S rRNA-c (MOTS-c)79, and small humanin-like peptides 1 to 6 (SHLPs 1–6)80 (Fig. 3). Humanin was firstly discovered MDP. It was identified during a search for the genes that lead to resistance to apoptosis in response to amyloid-β (Aβ) toxicity in the brains of Alzheimer’s disease (AD) patients. Professor Nishimoto coined the term “humanin” in reference to the hope of rescuing the “humanity” of patients. Since then, many researchers, including us81, have reported its powerful therapeutic effects in a wide range of diseases as a generic inhibitor of cell death77. In addition to humanin, Cobb et al. identified 6 putative mitochondrial sORFs in 16S rRNA and named them small humanin-like peptides75. The SHLP family members exhibit distinct expression patterns and play different biological roles77; for example, SHLP2 is involved in energy homeostasis82, SHLP3 is involved in cell survival80, and SHLP6 is involved in apoptosis80.

Mitochondria-derived peptides (MDPs) are a class of putative sORF-encoded polypeptides (SEPs) that are encoded within the mitochondrial ribosomal RNA (rRNA) region and have regulatory functions. The MDPs include humanin and SHLPs encoded by sORFs within the 16S rRNA and MOTS-c from the 12S rRNA region of the mitochondrial genome. MOTS-c, mitochondrial open reading frame of 12S rRNA-c; SHLPs, small humanin-like peptides.
Another MDP, MOTS-c, was identified as an exercise mimetic leading to systemic improvement of insulin sensitivity and metabolic homeostasis in obese mice79. As described above, MOTS-c was named for its sequence within the mitochondrial genome. Interestingly, if you flip the word ‘MOTS’ over, it looks like ‘SLOW’. Considering the regulatory role of MOTS-c in maintaining metabolic homeostasis, it is possible that MOTS-c can slow aging or treat age-related diseases in the context of mitochondrial signal transduction (see below). Overall, the discovery of MDPs may provide small but valuable pieces of the complex puzzle of the mitochondrial transcriptome. In that case, it is possible that mitochondria utilize their own proteins MDPs as representatives in signaling pathways.
Mitochondrial-encoded messengers in mitochondrial signal transduction
As endosymbionts, mitochondria retain a portion of the original bacterial genome that coevolved with the nuclear genome. Considering the putative energetic incentive for gene transfer, keeping small bioactive peptides in the mitochondrial genome as key messengers would be the most favorable means for primitive mitochondria to communicate with the nucleus.
MOTS-c is a novel signaling messenger that allows communication between mitochondria and the nucleus83 (Fig. 4). In response to metabolic and oxidative stress, MOTS-c displays translocation from the mitochondria to the nucleus. This distinct cellular localization pattern suggests that MOTS-c may play diverse roles based on its location. For example, MOTS-c in the nucleus can directly bind specific motifs, such as antioxidant response elements (AREs), which regulate the expression of a subset of genes involved in stress response and defense. Thus, MOTS-c is proposed to be a key component encoded by the mitochondrial genome that acts as a signaling molecule under stress. However, exactly how and when MOTS-c is released from mitochondria under stress remain unclear.

Mitochondrial MOTS-c can trigger mitochondria-to-nucleus signaling. MOTS-c, under stress conditions, is released from the mitochondria and translocated to the nucleus. The MOTS-c-activated signaling cascade can induce the expression of protective genes in a redox-dependent manner. MOTS-c, mitochondrial open reading frame of 12S rRNA-c.
Typically, mitochondria have been considered to be semi-autonomous, not fully autonomous, organelle which are tightly regulated by nuclear activity. As discussed above, a growing body of research indicates that mitochondria actually control nuclear functions mainly through retrograde signaling. Generally, cellular signal transduction employs input-to-output transformation16. In the same way, mitochondrial signal transduction appears to involve distinct cellular processes: sensing, integration, and signaling (see16 and references therein). MOTS-c seems to be the first identified signaling messenger encoded within the mitochondrial genome. In MOTS-c-mediated signaling, stressed mitochondria may sense a broad variety of inputs through receptors or transporters in the mitochondria. After the sensing process, these mitochondria may integrate information through sophisticated dynamics. From this perspective, a signaling molecule from stressed mitochondria can trigger powerful cell life or death signals depending on the integration of mitochondrial information. Asking how influential this process is, and what the underlying mechanisms are, is worthwhile.
Mitochondrial stress signaling influences organism health and diseases
The free radical theory of aging has brought mitochondria to the forefront of aging research84,85. Since then, mitochondrial dysfunction has been strongly implicated in a wide range of diseases, as well as aging. Based on the evidence presented earlier, the effect of certain stresses on mitochondria can link to defense mechanism against a host of diseases. Assessing how mitonuclear communication during stress regulates disease susceptibility and slows aging would be interesting43 (Table 1).
ROS signaling in longevity
Indeed, ROS can have both beneficial or detrimental effects on longevity depending on the conditions86. Interestingly, recent evidence clearly point to ROS as powerful mediators of longevity that act as downstream effectors of mitochondrial modulation during stress87. For instance, inhibiting respiration in C. elegans increases ROS levels, which leads to the upregulation of hypoxia-inducible factor 1 alpha (HIF‑1α) expression88. This HIF‑1α activation subsequently upregulates the expression of gene sets involved in the adaptive stress response and further increases lifespan. Remarkably, these pro-longevity effects seem to be dependent only on mitochondrial ROS, not cytoplasmic ROS89. Other researchers have come to similar conclusions on the impact of ROS in the context of mitonuclear communication. The nuclear response triggered by mitochondrial ROS under stress produces therapeutic benefits by activating a wide range of transcription factors, including NRF290, forkhead box O (FOXO)91, and heat shock factor 1 (HSF1)92, in various organisms. When blocking the generation of mitochondria under stress, the beneficial effects of stress resistance are significantly reduced93. Taken together, pro-oxidant agents could be putative therapeutic candidates for human health in some situations. Much remains to be learned about how our ROS respond to our S.O.S. signals in the context of mitohormesis, a coordinated adaptive stress response promoting longevity94,95.
Therapeutic effects of mitonuclear UPRmt signaling
Given the importance of the UPRmt in mitochondrial stress signaling, determining whether there is a direct link between the UPRmt and disease susceptibility would be interesting. Surprisingly, Tian et al. revealed a novel mechanism through which the UPRmt alters chromatin organization, ultimately extending the lifespan of C. elegans96. These effects are conserved in mice97 and humans98, indicating the existence of a conserved epigenetic system that can regulate biological aging through mitochondrial stress signaling. Interestingly, Aβ exposure in SH-SY5Y neuroblastoma cells triggers the UPRmt response. Inhibiting the UPRmt results in marked neurological deterioration99. This scientific evidence supports the use of novel therapeutic strategies involving mitohormetic signaling induced by mitochondrial stress.
Correlation does not imply causation. The profound impact of the UPRmt on health and lifespan should be considered from a systemic perspective. Our body is complicated, and there is no way around this fact. Like ROS, when these stress responses are continuously activated, it may also induce some detrimental outcomes. Furthermore, the UPRmt is inseparable from other mitochondrial stress signaling including ISR100. This complexity hinders the identification of the true starting and ending points of the UPRmt. Presently, the function of the UPRmt and interaction in mitochondrial diseases have gradually attracted attention101. Mitochondrial stress likely encompasses many different physiological scenarios. How the UPRmt is regulated during each scenario needs to be clarified for targeting mitochondrial function to treat diseases.
Mitonuclear MDPs in the stress response as therapeutic targets
As discussed above, mitochondrial DNA contains sORFs that encode functional polypeptides. The first category of biologically active peptides in mitochondrial sORFs could be MDPs. Since their discoveries, MDPs have been extensively studied in the context of aging (see78 and references therein). Multiple studies have contiuously suggested humanin and MOTS-c as biomarkers for various age-related diseases. Indeed, MDPs, such as humanin102 and MOTS-c103, have stress-responsive characteristics. In particular, MOTS-c activation by mitochondrial stress facilitates adaptation to stress and restores homeostasis through retrograde signaling83.
The devil is very much in the details. Considering similar characteristics of MDPs, there is a possibility that another MDP can act as a signaling molecule to control nuclear activity. Besides transcriptional regulation, nuclear gene expression is also controlled at the level of translation. As such, asking why all MDPs are encoded only in the rRNAs of mitochondrial genome is worthwhile. At present, there are no definitive answers. Evolutionarily, there is a growing perspective that ribosomes may have evolved to enhance translation and increase genetic code expansion in living cells104.
It is possible that primitive mitochondria might have retained specific ribosomal proteins within their own rRNA regions during endosymbiosis. For that strategy, mitochondria might keep small peptides within their own rRNA regions to 1) effectively reduce host-specific effects on synthetic mitochondrial genes and 2) translationally control gene expression in the host nucleus, specifically in the context of an orthogonal central dogma (see105 and references therein). We cannot yet tell with certainty whether MDPs can control the nuclear translational system, but this question needs to be answered.
Conclusion and future prospects
“Even when it leads you off the well-worn path, and that will make all the difference”
Steve Jobs (2005)106
Various aspects of mitochondrial evolution remain enigmatic. The problem may lie not in determining the details but rather in determining why bacterial endosymbionts became mitochondria. The endosymbiotic mitochondrion may have evolved a sophisticated communication system with the rest of the host cell, including the nucleus. In this mitochondria-centered communication, mitochondria might utilize diverse messengers or signals to restore and maintain mitochondrial and cellular functions in the ever-changing cellular milieu.
As emphasized by Steve Jobs106 (see above), there is a wealth of novel molecular biology evidence that seems unlikely to be connected, such as mitochondria-derived peptides, open reading frames, mitochondria-to-nucleus signaling, and mitochondrial stress. Will these dots connect in the future, and, if so, are we in a new era in which science and medicine will adopt a mitochondria-centric view? When that time comes, the mysteries of midi-chlorians in the Star Wars1 may unfold.
References
Lucas, G. Star Wars: Episode I—The Phantom Menaceed 20th Century Fox (1999).
Know, L. Mitochondria and the Future of Medicine: The Key to Understanding Disease, Chronic Illness, Aging, and Life Itself (Chelsea Green Publishing, 2019).
Woockieepedia. Midi-chlorian https://starwars.fandom.com/wiki/Midi-chlorian?veaction=edit§ion=6.
Detmer, S. A. & Chan, D. C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879 (2007).
Ono, T., Isobe, K., Nakada, K. & Hayashi, J. I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28, 272–275 (2001).
Chen, H. et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 (2010).
Twig, G. et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008).
Chandel, N. S. Evolution of mitochondria as signaling organelles. Cell Metab. 22, 204–206 (2015).
Archibald, J. M. Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 25, R911–R921 (2015).
Margulis, L. Origin of Eukaryotic Cells (Yale University Press, 1970).
Martin, W. F., Garg, S. & Zimorski, V. Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. Lond. B: Biol. Sci. 370, 20140330 (2015).
Martin, W. & Muller, M. The hydrogen hypothesis for the first eukaryote. Nature 392, 37–41 (1998).
Cavalier-Smith, T. The origin of eukaryotic and archaebacterial cells. Ann. N. Y Acad. Sci. 503, 17–54 (1987).
Baldauf, S. L., Palmer, J. D. & Doolittle, W. F. The root of the universal tree and the origin of eukaryotes based on elongation factor phylogeny. Proc. Natl Acad. Sci. USA 93, 7749–7754 (1996).
Gray, M. W., Burger, G. & Lang, B. F. Mitochondrial evolution. Science 283, 1476–1481 (1999).
Picard, M. & Shirihai, O. S. Mitochondrial signal transduction. Cell Metab. 34, 1620–1653 (2022).
Garrido, C. et al. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423–1433 (2006).
Liu, X., Kim, C. N., Yang, J., Jemmerson, R. & Wang, X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 (1996).
Skulachev, V. P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Asp. Med. 20, 139–184 (1999).
Atlante, A. et al. Cytochrome c is released from mitochondria in a reactive oxygen species (ROS)-dependent fashion and can operate as a ROS scavenger and as a respiratory substrate in cerebellar neurons undergoing excitotoxic death. J. Biol. Chem. 275, 37159–37166 (2000).
Huttemann, M. et al. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 11, 369–381 (2011).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Resta, G., Stramezzi, A. M., Sacco, R., Ciuffreda, M. & Casasco, A. [Fluoridation using iontophoresis. In vitro experimentation]. Prev. Assist Dent. 16, 35–38 (1990).
Fan, J., Krautkramer, K. A., Feldman, J. L. & Denu, J. M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 10, 95–108 (2015).
Moussaieff, A. et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402 (2015).
Cai, L., Sutter, B. M., Li, B. & Tu, B. P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437 (2011).
Tu, B. P., Kudlicki, A., Rowicka, M. & McKnight, S. L. Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science 310, 1152–1158 (2005).
Zhu, D. et al. NuRD mediates mitochondrial stress-induced longevity via chromatin remodeling in response to acetyl-CoA level. Sci. Adv. 6, eabb2529 (2020).
Jacob, F. & Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356 (1961).
Commoner, B., Townsend, J. & Pake, G. E. Free radicals in biological materials. Nature 174, 689–691 (1954).
Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 (2011).
Schulz, T. J. et al. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293 (2007).
Shadel, G. S. & Horvath, T. L. Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569 (2015).
Srinivasan, S. et al. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 35, 1585–1595 (2016).
English, J., Son, J. M., Cardamone, M. D., Lee, C. & Perissi, V. Decoding the rosetta stone of mitonuclear communication. Pharm. Res. 161, 105161 (2020).
Storz, P. Reactive oxygen species-mediated mitochondria-to-nucleus signaling: a key to aging and radical-caused diseases. Sci. STKE 2006, re3 (2006).
Slater, E. C. & Cleland, K. W. The effect of calcium on the respiratory and phosphorylative activities of heart-muscle sarcosomes. Biochem. J. 55, 566–590 (1953).
Rossi, A., Pizzo, P. & Filadi, R. Calcium, mitochondria and cell metabolism: a functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 1866, 1068–1078 (2019).
Giorgi, C., Marchi, S. & Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730 (2018).
Biswas, G. et al. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18, 522–533 (1999).
Luo, Y., Bond, J. D. & Ingram, V. M. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc. Natl Acad. Sci. USA 94, 9705–9710 (1997).
Berridge, M. J., Lipp, P. & Bootman, M. D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21 (2000).
Quiros, P. M., Mottis, A. & Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226 (2016).
Zimmerman, A. N. & Hulsmann, W. C. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature 211, 646–647 (1966).
Giorgi, C. et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium 52, 36–43 (2012).
Danese, A. et al. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenerg. 1858, 615–627 (2017).
Rimessi, A. et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy 9, 1677–1686 (2013).
Dolezal, P., Likic, V., Tachezy, J. & Lithgow, T. Evolution of the molecular machines for protein import into mitochondria. Science 313, 314–318 (2006).
Pellegrino, M. W., Nargund, A. M. & Haynes, C. M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416 (2013).
Sweeney, P. et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegener. 6, 6 (2017).
Martinus, R. D. et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem 240, 98–103 (1996).
Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013).
Haynes, C. M. & Ron, D. The mitochondrial UPR - protecting organelle protein homeostasis. J. Cell Sci. 123, 3849–3855 (2010).
Fiorese, C. J. et al. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043 (2016).
Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 16, 81 (2018).
Hamacher-Brady, A. & Brady, N. R. Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol. Life Sci. 73, 775–795 (2016).
Kafri, M., Metzl-Raz, E., Jona, G. & Barkai, N. The cost of protein production. Cell Rep. 14, 22–31 (2016).
Costa-Mattioli, M. & Walter, P. The integrated stress response: From mechanism to disease. Science 368 (2020).
Wang, F., Zhang, D., Zhang, D., Li, P. & Gao, Y. Mitochondrial protein translation: emerging roles and clinical significance in disease. Front Cell Dev. Biol. 9, 675465 (2021).
Young, S. K. & Wek, R. C. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 291, 16927–16935 (2016).
Fritsch, C. et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 22, 2208–2218 (2012).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Harding, H. P. et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 (2000).
Vattem, K. M. & Wek, R. C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl Acad. Sci. USA 101, 11269–11274 (2004).
Lu, P. D., Harding, H. P. & Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33 (2004).
Zhou, D. et al. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 283, 7064–7073 (2008).
Palam, L. R., Baird, T. D. & Wek, R. C. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 286, 10939–10949 (2011).
Yang, Y. & Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 11, 911–919 (2019).
Ryoo, H. D. & Vasudevan, D. Two distinct nodes of translational inhibition in the integrated stress response. BMB Rep. 50, 539–545 (2017).
Wright, B. W., Yi, Z., Weissman, J. S. & Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 32, 243–258 (2022).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Hao, Y. et al. SmProt: a database of small proteins encoded by annotated coding and non-coding RNA loci. Brief. Bioinform 19, 636–643 (2018).
Brunet, M. A. et al. OpenProt 2021: deeper functional annotation of the coding potential of eukaryotic genomes. Nucleic Acids Res 49, D380–D388 (2021).
Neville, M. D. C. et al. A platform for curated products from novel open reading frames prompts reinterpretation of disease variants. Genome Res. 31, 327–336 (2021).
Saghatelian, A. & Couso, J. P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 11, 909–916 (2015).
Ma, J. et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J. Proteome Res. 13, 1757–1765 (2014).
Miller, B., Kim, S. J., Kumagai, H., Yen, K. & Cohen, P. Mitochondria-derived peptides in aging and healthspan. J. Clin. Invest. 132 (2022).
Hashimoto, Y. et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl Acad. Sci. USA 98, 6336–6341 (2001).
Lee, C. et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454 (2015).
Cobb, L. J. et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY) 8, 796–809 (2016).
Kim, K. H. Intranasal delivery of mitochondrial protein humanin rescues cell death and promotes mitochondrial function in Parkinson’s disease. Theranostics 13, 3330–3345 (2023).
Kim, S. K. et al. Mitochondria-derived peptide SHLP2 regulates energy homeostasis through the activation of hypothalamic neurons. Nat. Commun. 14, 4321 (2023).
Kim, K. H., Son, J. M., Benayoun, B. A. & Lee, C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 28, 516–524 e517 (2018).
Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 (1956).
Liu, X. et al. Mitochondria as a sensor, a central hub and a biological clock in psychological stress-accelerated aging. Ageing Res. Rev. 93, 102145 (2024).
Shields, H. J., Traa, A. & Van Raamsdonk, J. M. Beneficial and detrimental effects of reactive oxygen species on lifespan: a comprehensive review of comparative and experimental studies. Front. Cell Dev. Biol. 9, 628157 (2021).
Mottis, A., Herzig, S. & Auwerx, J. Mitocellular communication: shaping health and disease. Science 366, 827–832 (2019).
Lee, S. J., Hwang, A. B. & Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136 (2010).
Schaar, C. E. et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 11, e1004972 (2015).
Rushmore, T. H., Morton, M. R. & Pickett, C. B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 266, 11632–11639 (1991).
Owusu-Ansah, E., Yavari, A., Mandal, S. & Banerjee, U. Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 40, 356–361 (2008).
Yang, W. & Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447 (2010).
Sena, L. A. & Chandel, N. S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167 (2012).
Yun, J. & Finkel, T. Mitohormesis. Cell Metab. 19, 757–766 (2014).
Barcena, C., Mayoral, P. & Quiros, P. M. Mitohormesis, an antiaging paradigm. Int. Rev. Cell Mol. Biol. 340, 35–77 (2018).
Tian, Y. et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208 (2016).
Merkwirth, C. et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223 (2016).
Suomalainen, A. & Battersby, B. J. Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 19, 77–92 (2018).
Sorrentino, V. et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552, 187–193 (2017).
Lu, H. et al. Mitochondrial unfolded protein response and integrated stress response as promising therapeutic targets for mitochondrial diseases. Cells 12 (2022).
Melber, A. & Haynes, C. M. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295 (2018).
Woodhead, J. S. T. et al. High-intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J. Appl Physiol. 128, 1346–1354 (2020).
Reynolds, J. C. et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 12, 470 (2021).
Liu, F., Bratulic, S., Costello, A., Miettinen, T. P. & Badran, A. H. Directed evolution of rRNA improves translation kinetics and recombinant protein yield. Nat. Commun. 12, 5638 (2021).
Liu, C. C., Jewett, M. C., Chin, J. W. & Voigt, C. A. Toward an orthogonal central dogma. Nat. Chem. Biol. 14, 103–106 (2018).
AggiEnglish. Steve Jobs Stanford Commencement Speech 2005 https://aggienglish.tistory.com/56 (2020).
Eisenberg, T. et al. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 19, 431–444 (2014).
Schmeisser, S. et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell 12, 508–517 (2013).
Heidler, T., Hartwig, K., Daniel, H. & Wenzel, U. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology 11, 183–195 (2010).
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M. & Haynes, C. M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 (2012).
Acknowledgements
This work was supported by a grant funded by the National Research Foundation of Korea (RS-2023-00243300) to K.H.K.
Author information
Authors and Affiliations
Contributions
The authors confirm that they contributed to the paper as follows: C.B.L. contributed to drafting the figure. K.H.K. conceptualized the review, collected the information, generated the figures and tables, and wrote and revised the manuscript. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, K.H., Lee, C.B. Socialized mitochondria: mitonuclear crosstalk in stress. Exp Mol Med (2024). https://doi.org/10.1038/s12276-024-01211-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s12276-024-01211-4
