
Abstract
Great advances in deep learning have provided effective solutions for prediction tasks in the biomedical field. However, accurate prognosis prediction using cancer genomics data remains challenging due to the severe overfitting problem caused by curse of dimensionality inherent to high-throughput sequencing data. Moreover, there are unique challenges to perform survival analysis, arising from the difficulty in utilizing censored samples whose events of interest are not observed. Convolutional neural network (CNN) models provide us the opportunity to extract meaningful hierarchical features to characterize cancer subtype and prognosis outcomes. On the other hand, feature selection can mitigate overfitting and reduce subsequent model training computation burden by screening out significant genes from redundant genes. To accomplish model simplification, we developed a concise and efficient survival analysis model, named CNN-Cox model, which combines a special CNN framework with prognosis-related feature selection cascaded Wx, with the advantage of less computation demand utilizing light training parameters. Experiment results show that CNN-Cox model achieved consistent higher C-index values and better survival prediction performance across seven cancer type datasets in The Cancer Genome Atlas cohort, including bladder carcinoma, head and neck squamous cell carcinoma, kidney renal cell carcinoma, brain low-grade glioma, lung adenocarcinoma (LUAD), lung squamous cell carcinoma, and skin cutaneous melanoma, compared with the existing state-of-the-art survival analysis methods. As an illustration of model interpretation, we examined potential prognostic gene signatures of LUAD dataset using the proposed CNN-Cox model. We conducted protein–protein interaction network analysis to identify potential prognostic genes and further analyzed the biological function of 13 hub genes, including ANLN, RACGAP1, KIF4A, KIF20A, KIF14, ASPM, CDK1, SPC25, NCAPG, MKI67, HJURP, EXO1, HMMR, whose high expression is significantly associated with poor survival of LUAD patients. These findings confirmed that CNN-Cox model is effective in extracting not only prognosis factors but also biologically meaningful gene features. The codes are available at the GitHub website: https://github.com/wangwangCCChen/CNN-Cox.
Similar content being viewed by others


Introduction
Cancer is a heterogeneous disease driven by diverse gene mutations, and the analysis of genomics data is essential to extract molecular factors related to disease progression and prognosis1. A large amount of various omics data has been generated by high-throughput sequencing techniques, such as genomics, transcriptomics, proteomics, and metabolomics. There are some prominent resources of cancer genomics data, such as The Cancer Genome Atlas (TCGA)2, the Catalog of Somatic Mutations in Cancer3, and the Molecular Taxonomy of Breast Cancer International Consortium4. The main prediction tasks in the biomedical field include cancer diagnosis, tumor subtype classification, and prognosis prediction5,6,7,8. Predicting cancer prognosis accurately from large-scale genomics data remains challenging due to the complexity of genomics data. Among tens of thousands of genes, most genes do not contain informative mutations, making it critical to extract prognosis-related key gene features7. In addition, there are unique challenges to perform survival analysis, arising from the difficulty in utilizing censored samples whose events of interest are not observed.
Survival analysis methods can be classified into two main categories: statistical methods and machine learning-based methods9. Cox regression model is the most widely used statistical method, which is built on the proportional hazards assumption and partial likelihood for parameter estimation10. There are some variants of Cox model in the literature, such as regularized Cox models with l1-norm, l2-norm or elastic-net penalty, CoxBoost, and time-dependent Cox model11,12. Machine learning based survival analysis methods are usually applied to high-dimensional problems and take advantage of optimization to learn the nonlinear relation between covariates and survival time. Survival trees, Bayesian methods, support vector machines, and neural networks are the most prevalent machine learning-based methods for survival analysis13,14,15,16,17,18.
Deep learning technologies have achieved great success in computer vision field, with advantages of learning nonlinear low-dimensional representations, such as convolutional neural network (CNN), auto-encoders, and recurrent neural networks19,20,21,22. Specially for high-throughput genomics data, deep learning has been confirmed to be able to capture biologically relevant features from high-dimensional genomics data23,24,25,26. Several promising studies have applied variational auto-encoders on gene expression data for cancer subtype classification and survival analysis25,26. Deep learning approaches usually employ multi-layer neural networks, with huge numbers of parameters to be optimized. Optimizing large number of parameters with limited samples tends to cause the overfitting problem that leads to ineffective performance on the test data.
CNN architecture uses convolution filters to automatically extract high-level features from raw elements, enabling the network trained much deeper with fewer parameters by weight sharing and local connections27. However, the application of CNN model on genomics data still has limitations, because gene expression data lacks local motifs and can’t show spatial coherence like image data. Lyu and Haque proposed to transform gene expression vectors into images based on the chromosome location, and subsequently applied CNN models for tumor type classfication28. Ma and Zhang presented OmicsMapNet approach to rearrange omics data into structured images where functional related molecular features are spatially adjacent. Then they trained CNN models on RNA-seq data to predict the malignancy grade of diffuse gliomas29. Guillermo proposed to rearrange RNA-seq data into gene expression images using gene relative positions based on their molecular function. To address the overfitting problem, they adopted the transfer learning approach to first pre-train CNN model on non-lung TCGA Pan-Cancer samples, and the resulting network was subsequently fine-tuned on lung cancer samples to improve survival prediction of lung cancer patients30.
As is known to all, training CNN model on genomics data involves the overfitting problem resulting from the curse of dimensionality inherent to gene expression data. Several studies have shown that shallower CNN models are more effective in cancer genomics prediction, by reducing the number of training parameters to mitigate the overfitting problem24. Hence, feature selection approaches should be attached importance to analyzing genomics data. To effectively identify prognosis-associated genes, Shin and Park proposed a novel neural network-based feature selection algorithm named cascaded Wx (CWx), which ranks features based on the capability of distinguishing high-risk and low-risk groups in a cascaded manner31. The results indicated that CWx identified the best candidate gene set to predict survival prognosis, highlighting CWx algorithm as an effective feature selection approach in survival analysis.
The main objective of this work is to present a new CNN-based survival analysis model that combines special 1D-CNN designs with prognosis-related feature selection CWx approach, with the advantage of superior performance and computation efficiency with light training parameters. To evaluate the effectiveness of the newly proposed method, we conduct extensive experiments on TCGA RNA-seq expression datasets from seven representative cancer types, compared with the existing state-of-the-art survival analysis methods. The results demonstrated that the newly proposed method achieved more stable and superior survival prediction accuracy assessed by the concordance index. Furthermore, effective feature selection allows us to perform model interpretations to elucidate prognosis gene markers for each cancer type.
Materials and methods
Dataset and preprocessing
In this study, we used the public TCGA pan-cancer RNA-seq dataset, which can be accessed by the UCSC Xena data browser (https://xenabrowser.net/datapages/)32. The dataset contains 10,535 samples from 33 tumor types, measured by log2(TPM + 0.001) transformed RSEM values, in which the number of original genes is 60,498. We firstly retained the top 20K most variably expressed genes based on the median absolute deviation, and removed genes with low information burden (mean <0.5 or standard deviation <0.8). A total of 6407 genes remained after the filtering step. The clinical outcome variables are derived from the Pan-cancer Atlas phenotype dataset, with four types of survival endpoints, overall survival, disease-specific survival, disease-free interval, and progression-free interval.
In this study, we chose overall survival as the survival endpoint. We denote gene features as X∈RN×p, survival time as T∈RN, binary event indicator as δ∈RN, N is the number of patients and p is the number of gene features. If δi = 1, Ti represents the survival time between the start of observation and occurrence of event (death). If δi = 0, Ti represents the censored time between the start and the end of observation. For each gene feature Xi,j, we calculated normalized Z-score by subtracting mean \(\overline {X_j}\) and dividing it by the standard deviation σj of gene j across all samples, that is, \(Z_{i,j} = \frac{{X_{i,j} - \overline {X_j} }}{{\sigma _j}}\). We selected seven different cancer types, bladder carcinoma (BLCA), head and neck squamous cell carcinoma (HNSC), kidney renal cell carcinoma (KIRC), brain low-grade glioma (LGG), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and skin cutaneous melanoma (SKCM), since they have more than 400 patient samples and 50 uncensored samples. Table 1 provides the sample information of each cancer type used in the study. Due to survival differences of different cancer type patients, referring to previous literature on survival analysis, we chose to fit models for each cancer type separately.
CNN-Cox model combined with CWx feature selection
For a given sample i, it is represented by a triplet (xi,yi,δi), xi∈R1×p is the feature vector, δi is the event indicator, i.e., δi = 1 represents an occurred event (death) and yi is time to event Ti for an uncensored instance, otherwise δi = 0 represents a censored instance and yi is the censored time Ci. The target of survival analysis is to estimate the survival time Tj for a new sample j with gene feature xj. The most common survival analysis model is the Cox proportional hazards (CoxPH) model, following the proportional hazards assumption:
The partial likelihood is the product of the probability of all samples, defined as follows:
where Ri is the set of patients still at risk of death at any time t which is larger than Ti of the ith subject, i.e., Ri = {j:Tj > Ti}. The coefficient vector β is estimated by maximizing the partial likelihood, or equivalently, minimizing the negative log-partial likelihood10:
Faraggi and Simon18 extended CoxPH model to nonlinear neural network framework, replacing the log hazard ratio βTxi in CoxPH model by the output of neural network g(xi,ω). Therefore, the nonlinear hazard function becomes \(h(t,x_i) = h_0(t) \cdot {{{{{\rm{exp}}}}}}(g(x_i,\omega ))\), and the negative log-partial likelihood becomes
However, this simple extended model is not feasible for high-throughput gene expression data.
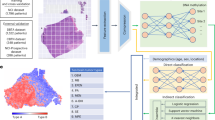
Different CNN models have been proposed to apply for cancer subtype classification tasks on gene expression data33,34,35. Mostavi proposed three simplified CNN designs with only one convolution layer directly trained on unstructured gene features, named 1D-CNN, 2D-Vanilla-CNN, and 2D-Hybrid-CNN respectively33. The 2D-Vanilla-CNN model follows the common CNN framework, applies 2D convolution kernels on image inputs to extract local features, and passes the output to a max-pooling layer, a fully connected layer, and a prediction layer. Inspired by parallel towers in Resnet module, 2D-Hybrid-CNN model applies two 1D convolution kernels, one with the size of a row slides vertically and the other one with the size of a column slides horizontally across the 2D matrix input (see Fig. 1A). For the 1D-CNN model, it takes the gene expression as a vector and applies one-dimensional convolution kernels to the input vector (see Fig. 1B). It is noteworthy that gene features in the vectorized input are arranged in the gene symbol’s alphabetic order from the data file, and we did not make a specific permutation of the gene positions. The 1D-CNN model captures temporal relationships between adjacent input values, yet 2D-Hybrid-CNN model can capture global unstructured features. The 2D-Vanilla-CNN model is not only highly-intensive trained and more difficult to converge, but also achieved lower prediction accuracy comparing with the other two CNN model designs. Hence, we chose to develop new survival analysis models based on simpler 1D-CNN and 2D-Hybrid-CNN architectures.

A CNN-Cox model based on 2D-Hybrid-CNN architecture. B 1D-CNNCox model based on 1D-CNN architecture. C Workflow for preprocessing, CWx feature selection, training, and testing of CNN-Cox model.
Inspired by CNN-based cancer type classification models outlined in Ref. 33, we extended it to survival analysis and designed a similar CNN framework for survival analysis models: CNN-Cox model based on 2D-Hybrid-CNN framework, and 1D-CNNCox model based on 1D-CNN framework. In this study, we proposed a novel survival analysis model that takes advantage of the CNN and Cox proportional hazards model, through performing an output Cox-regression layer based on activation levels of the hidden layer of the CNN framework. Hence, our proposed CNN-Cox model architecture is a combination of 2D-Hybrid-CNN and CoxPH model, as illustrated in Fig. 1A. The objective function of CNN-Cox is the negative partial log-likelihood defined at Eq. (4), with nonlinear proportional hazards g(xi,ω) defined as follows:
wh and wv denote horizontal and vertical 1D convolution kernels, respectively. MaxPool denotes max-pooling layer, Fl denotes flatten layer, wf and bf denote the weights and bias of full connected layer, β and d denote the weights and bias of Cox-regression output layer. Accordingly, the nonlinear proportional hazards g(xi,ω) of 1D-CNNCox model is defined as follows, shown in Fig. 1B:
Feature selection is an important dimension reduction method, extremely useful for genomics data analysis tasks. Park et al.31 developed a neural network-based feature selection method named Wx, which ranks features based on the discriminative index score to distinguish different groups. They further proposed a prognosis-related feature selection algorithm named cascaded Wx (CWx), which ranks gene features based on the discriminative index score to classify high-risk and low-risk groups with different survival time cutoffs in a cascade manner. Specifically, the top features are selected using the following discriminating power equation:
Whigh denotes training weights linked to high-risk output, Wlow denotes weights linked to low-risk output of the final layer. \(\bar X_{j,{{{{{\rm{high}}}}}}}\), \(\bar X_{j,{{{{{\rm{low}}}}}}}\) represent average expression values of gene j in the high-risk and low-risk groups, respectively. Firstly, patient samples were divided into high-risk and low-risk groups according to whether they have survived for S years, that is, dead patients within S years form the high-risk group, whereas patients who lived more than S years form the low-risk group. The censored patients were excluded in the training stage. The cascade second and third steps are similar as the first step with different survival time cutoffs (S1 versus S2, S3 versus S4). Meanwhile, input gene features are reduced by a quarter after each step, retaining one quarter of top genes in sorted scores in descending order, as illustrated in Fig. 1C. The evaluation revealed that cascade framework significantly improved prognostic-related feature selection performance. Motivated by the success of CWx algorithm, we develop a novel CNN-based survival analysis approach, integrating CNN-Cox models with CWx feature selection to improve survival prediction performance. The workflow for our proposed survival analysis model is shown in Fig. 1C. For the preprocessed gene expression data of 7 different cancer types with 6407 genes, the CWx feature selection approach was first applied to select different numbers of prognostic gene features (3000/2000/1000/500/196/144/100/81/49/25), and then CNN-Cox model was trained and evaluated on datasets with selected gene features using the five-fold cross-validation strategy based on optimal hyper-parameters selected on independent validation data subsets.
Evaluation metrics
To evaluate the survival prediction performance of all models, we used the five-fold cross-validation strategy shown in Fig. 1C. In each random sampling, we trained the models with 80% of the data, and the remaining 20% was used for evaluating models. The prediction performance in survival analysis was evaluated using C-index, which is the concordance index to measure concordance between predicted risk and actual survival outcome15. The C-index can be seen as a summation over relative risk of all events, where patients with longer survival time and lower log hazard ratios, or patients with shorter survival time and higher log hazard ratios are considered concordant. The C-index is computed as follows:
Where m denotes the number of all comparable pairs, \(\hat c\) is the C-index score value between 0 and 1. We also calculated the micro-average C-index on seven different cancer type datasets, defined as follows:
where ni is the number of samples in the ith cancer type, \({{{\hat{ c}}}}_{{{{{{i}}}}}}\) is the predicted C-index value on the ith cancer type dataset.
Results
Hyper-parameter selection
With the aim of assessing the effectiveness of CNN-Cox model architectures, we first compared new models with Cox-ElasticNet (Cox-EN) and standard neural network-based NN-Cox model, which has two fully connected hidden layers and an output Cox-regression layer. We implemented neural network-based models using Keras with TensorFlow backend, Cox-EN model using scikit-survival package. For CNN-Cox model with 2D-Hybrid-CNN structure, we reshaped the screened 6407 gene inputs as a matrix with 100 rows and 65 columns by adding 93 zeros in the last column.
For a fair comparison, the hyper-parameters of each model were optimized using the grid search method through the five-fold cross-validation on the training data subsets for each cancer type. The hyper-parameters of CNN-Cox model include the size of 1D convolution kernels (1st_CNN and 2nd_CNN), the number of nodes in the fully connected layer (dense_size). The search ranges of three hyper-parameters in the grid search was respectively set as [8,16,32,64,128], [8,16,32,64,128] and [16,32,64,128,512]. For the 1D-CNNCox model, there are only two hyper-parameters, the size of 1D convolution kernel (CNN_size) and the number of nodes of the fully connected layer (dense_size). For the NN-Cox model, hyper-parameters include the number of nodes in two fully connected layers. For the Cox-EN model, it combines ℓ1 and ℓ2 penalties to perform feature selection, there is a hyper-parameter ℓ1-ratio which controls the regularization level. Supplementary Table S1 shows optimal hyper-parameters selected by the grid search method for four survival analysis models on seven cancer type datasets, respectively based on the original 6407 genes and 100 genes selected by the CWx approach.
As a demonstrating example, we plotted hyper-parameter selection process graphs for CNN-Cox model on the LGG dataset with 6407 genes in Supplementary Fig. S1. We can see that optimal hyper-parameter setup is (64,128,512), which is consistent with optimal hyper-parameter results in Supplementary Table S1.
Effectiveness of CNN-Cox survival analysis model
To further assess the effectiveness of the proposed CNN-Cox models, we made a comparison with five state-of-the-art survival models, including NN-Cox, Cox-EN, random survival forest (RSF), gradient boosting machines (GBM), survival support vector machine (SSVM), which are implemented by scikit-survival package and evaluated on seven cancer types, BLCA, SKCM, KIRC, LGG, HNSC, LUAD, and LUSC. The performance of C-index values of seven models on seven cancer type datasets based on a different number of genes in five times five-fold cross-validation are compared and shown in Supplementary Table S2, and Wilcoxon signed-rank test results of CNN-Cox model comparing with other baseline models on each dataset are shown in Fig. 4E. We can see that CNN-Cox model shows significantly better performance, except 1D-CNNCox and RSF model. Moreover, Table 2 listed the micro-average C-index values of all models for seven cancer type datasets, based on a different number of genes selected by CWx approach (6407, 3000, 2000, 1000, 500, 196, 144, 100, 81, 49, 25).
It can be seen from Table 2 that CNN-Cox and 1D-CNNCox outperform other models consistently in most cases, even using the original 6407 genes without feature selection. The average improvement of CNN-Cox against other models is nearly 2%, except the competitive performance of RSF model. Moreover, we performed Friedman test and post-hoc Bonferroni–Dunn test on these micro-average C-index values, assessing the statistical significance of the model improvement of CNN-Cox and 1D-CNNCox36. The significance value FF = 9.7206 is far greater than the critical value 2.2541 at α = 0.05 significance level, showing that these seven models perform significantly differently. Then we performed the post-hoc Bonferroni–Dunn test for paired comparisons of CNN-Cox against other baseline models. The critical difference (CD) diagram for statistical test results is shown in Table 2, where values on x axis denote average ranks of models. If the rank difference between two methods is smaller than CD = 2.490, the performance difference is not significant (connected by a horizontal line). We can see from CD diagram that CNN-Cox shows significantly better performance than other models, except 1D-CNNCox and RSF model.
We also plotted box-plots of C-index distributions for each cancer type in Fig. 2A, B. We can see that CNN-Cox and 1D-CNNCox (red and blue) both show superior performance on five cancer types, except KIRC and LUAD. We can see from Fig. 2B that these two models still keep superior performance on almost all seven datasets based on 100 genes selected by CWx, confirming the effectiveness of CWx feature selection for survival analysis models. In order to further verify the effectiveness of CNN-Cox network structure, we compared micro-average C-index values on seven datasets for each model based on a different number of genes selected by CWx approach shown in Fig. 2C. We can see that CNN-Cox and 1D-CNNCox (blue and orange) consistently achieved higher micro-average C-indexes than other baseline models. These results show the robustness and superiority of CNN-Cox and 1D-CNNCox models on survival prediction performance.

A Box-plot of C-index values using 6407 genes. B Box-plot of C-index values using 100 genes selected by CWx. C Micro-average C-index on seven datasets using a different number of genes selected by CWx. D C-index value difference between 6407 and 100 selected genes on each dataset.
From another point of view, we plotted C-index values difference between 100 genes and initial 6407 genes on each cancer type for each model shown in Fig. 2D. We can see that all models achieved positive C-index difference values, except Cox-EN and SSVM models on the KIRC and LGG datasets. These results further confirmed the effectiveness of CWx feature selection for survival prediction models, as it mitigates the overfitting problem on high-dimensional gene expression data. Hence, it revealed that CWx feature selection is very useful to learn meaningful prognosis-related gene signatures and further improve the survival prediction performance.
We also performed further survival analysis to evaluate the performance of CNN-Cox model in survival prediction. We divided patient samples for each cancer type into high-risk and low-risk groups based on their predicted hazard ratios. When the predicted hazard ratio is higher than the median hazard ratio of all patient samples, the sample is divided into the high-risk group; otherwise, it will be included in the low-risk group26. Figure 3 shows Kaplan–Meier plots and log-rank test results of high-risk and low-risk groups for seven different cancer types using CNN-Cox model. We can see that log-rank test p-values are lower than 0.001 and samples of different cancer types are divided into high-risk and low-risk groups significantly, except HNSC, LUAD, and LUSC. These survival analysis results revealed that CNN-Cox model can effectively split samples of different cancer types into high-risk and low-risk groups.

The patient samples are divided into high-risk and low-risk groups based on the predicted hazard ratios. A BLCA, B HNSC, C KIRC, D LGG, E LUAD, F LUSC, and G SKCM.
Inspired by the idea of utilizing clinical information, we used for reference the research work of Hao et al. on pathway-based sparse deep neural network model, named Cox-PASNet, to integrate genomics and clinical data for survival analysis37. High-dimensional genomics data would dominate the integration if it is combined with clinical data directly, due to the unbalanced size between them. Hence, we introduce clinical data to the model through a separate clinical layer. The effects of genomics data are captured by two parallel convolutional layers, whereas the clinical data are directly introduced into the output layer, along with the highest-level representation of the last hidden layer as shown in Fig. 4A. We chose three clinical characteristics (age at diagnosis, sex, stage at diagnosis) for six different cancer types, BLCA, HNSC, KIRC, LUSC, LUAD, and SKCM, since LGG has a large number of missing data on the cancer stage feature. The performance of C-index values of seven models on six cancer type datasets integrating 6407/100 genes and three clinical features are also compared and shown in Supplementary Table S2. We can see from Fig. 4B that CNN-Cox model shows better performance after the introduction of clinical layer, no matter when it is integrated with 6407 or 100 genes.

A Network architecture of CNN-Cox model adding a separate clinical layer. B Performance comparison of CNN-Cox model before and after adding the clinical layer on six datasets. C Comparison of running time of six models based on 6407 genes on seven datasets. D Comparison of running time of seven models based on 100 genes on seven datasets. E Wilcoxon signed-rank test results of CNN-Cox model comparing with other baseline models.
In addition, we made the comparison of running time for seven survival analysis models on seven cancer type datasets based on original 6407 and 100 selected genes, respectively, which is shown in Supplementary Table S3 and Fig. 4C, D. In the situation of high-dimensional 6407 genes, the running time is sorted in descending order: RSF > GBM > SSVM > 1D-CNNCox > CNN-Cox > NN-Cox > Cox-EN, especially the running time of RSF is dozens of times of CNN-Cox. In the situation of 100 genes, GBM, SSVM, and Cox-EN are more efficient than CNN-Cox, but the running time of RSF is still ten times of CNN-Cox. Although CNN-Cox shows a comparable and not-so-significant performance advantage over RSF, computational efficiency is one advantage of CNN-Cox over RSF.
Discussion
Identifying biologically meaningful gene subset is an essential step in discovering underlying mechanisms of cancer diseases. As an illustration of model interpretation for CNN-Cox survival analysis model, we investigated prognosis-related gene signatures for the LUAD dataset. Firstly, we conducted the gene set enrichment analysis (GSEA) to screen differential genes between the high-risk and low-risk groups.
GSEA and protein–protein interaction network analysis
GSEA is a method for assessing whether a fixed gene set shows statistically significant and concordant differences between two biological states (http://www.gsea-msigdb.org/gsea/)38. We performed GSEA analysis on the LUAD dataset with 6407 genes of 206 patient samples, attained by removing censored and missing samples on survival time. These samples were divided into high-risk and low-risk groups, by taking a 3-year survival time as a cutoff. Then we loaded gene sets files, phenotype labels, gene expression, and chip annotation files into GSEA software, with the adjusted p value FDR < 0.25 is set as the statistical significance cutoff level. We used the HALLMARK gene sets file from MSigDB39 (http://www.gsea-msigdb.org/gsea/msigdb/) as predefined gene sets.
The enrichment score (ES) in the GSEA analysis reflects the degree to which a gene set is over-represented at the top or bottom of a ranked list of genes. The top enriched gene set of the LUAD dataset with 6407 genes for the high-risk phenotype is HALLMARK_HYPOXIA, and the enrichment plot is shown in Fig. 5A. The top plot shows the running ES as walking down the ranked list, the score at the peak is the ES for the gene set, and the leading edge subset of the gene set contains 70 genes that contribute most to ESs. As the statistic for accounting for correlations between gene set and expression data, the normalized ES of HYPOXIA gene set is 1.86978 with statistical significance nominal p value P = 0.004219 and adjusted p value FDR = 0.111392. The heatmap of top 50 features for each phenotype is shown in Fig. 5B, where red colors denote high expressed, blue colors denote low expressed between ranked genes and phenotype. For the LUAD dataset, we achieved 1072 differential genes whose ESs are less than −0.12 or greater than 0.15 in the GSEA analysis.

A Enrichment plot of 6407 genes for the high-risk phenotype of LUAD dataset. B Heatmap of top 50 genes for each phenotype of LUAD dataset. C Hub genes of LUAD. D Hub genes of LUSC. E Hub genes of BLCA. F Hub genes of KIRC. G Hub genes of LGG. H Hub genes of SKCM. I Hub genes of HNSC.
As we know, PPI play an essential role in regulating biological processes. The densely connected regions in PPI network may serve as enriched function clusters. The 59 overlapping genes are obtained by the intersection of 1072 differential genes with 100 genes selected by CWx method. We imported these 59 genes into STRING database to construct PPI network (https://string-db.org/)40, resulting 56 nodes and 42 edges when the confidence score threshold was set as 0.9. We used Markov cluster algorithm in STRING to identify function clusters of PPI network. The most significant cluster contains 13 hub genes, including ANLN, RACGAP1, KIF4A, KIF20A, KIF14, ASPM, CDK1, SPC25, NCAPG, MKI67, HJURP, EXO1, and HMMR, as shown in Fig. 5C. For the other six cancer type datasets, we also conducted GSEA and PPI network analysis to identify hub genes for each cancer type dataset, which are respectively shown in Fig. 5D–I. We can see that APOH, FGA, FGG, HPX, ITGAX, SERPIND1 are hub genes of LUSC dataset. CD247, CD3D, CD3G, CD5, EPHA4, IKZF3, LCK, SLA2, SRC ZAP70 are hub genes of BLCA dataset. ADH6, ALDH3A2, BBOX1, GATM are hub genes of KIRC dataset. CCNB1, GADD45A, LPIN3, NUF2, PKMYT1, PTTG1, WEE1 are hub genes of LGG dataset. BST2, GBP2, IFIT3, IRF1, PSMB8, PSMB9, STAT1 are hub genes of SKCM dataset. CCL20, CSF3, CXCL2, CXCL8, IL1B, KITLG are hub genes of HNSC dataset.
Biological functions of hub genes
Hub genes are highly interconnected genes and play central roles in the PPI network. They may be potential biomarkers and therapeutic targets. To determine the biological functions of these 13 hub genes, we used Gene Ontology (GO) analysis (https://david.ncifcrf.gov/) to identify enriched genes using the statistical significance threshold FDR < 0.05. Table 3 shows the most enriched GO biological processes terms in hub genes of PPI network for six cancer type datasets. The significant biological processes are enriched in mitotic cytokinesis, microtubule-based movement, mitotic nuclear division, and cell division.
In the most significant cluster 1, Anillin (ANLN) encodes an actin-binding protein that plays key roles in cell growth and migration in cytokinesis. Previous studies have confirmed that ANLN expression is associated with patient prognosis with the breast, bladder, and colorectal cancers. There are some evidence showing that ANLN is related to metastasis in LUAD by promoting epithelial-mesenchymal transformation of tumor cells41.
Rac GTPase activating protein 1 (RACGAP1) plays an essential role in the inducing of cytokinesis and promoting cancer proliferation and growth. RACGAP1 expression is significantly upregulated in pan-cancers, and high RACGAP1 expression is correlated with the poor prognostic outcome in six cancer types, including BRCA, LUAD, LGG, LAML, HNSC, and PAAD42.
Kinesin superfamily (KIF) comprises a group of microtubule-based and ATP-powered motor proteins, which participate in mitosis, intracellular transportation, and cytoskeletal reorganization. KIF4A has been identified as an oncogene and contributor to malignant progression in lung cancer, oral cancer, prostate cancer, and breast cancer43. The study observed that KIF4A expression is correlated with cancer stage, metastasis, and tumor dimension, and high KIF4A expression is significantly associated with shorter overall survival in multiple cancer types. KIF20A is a member of the kinesin superfamily-6, which localized at Golgi apparatus and participates in organelle dynamics. Previous studies have also shown that high KIF20A expression is associated with poor prognostic outcomes in pan-cancers, such as pancreatic, breast, glioma, prostate, and bladder cancers44. A similar oncogenic function of KIF14 in the cell cycle and proliferation has also been reported. Growing evidence showed that KIF family genes affect patients' prognosis outcomes by involving cell cycle-related biological processes and pathways45.
Abnormal spindle-like microcephaly-associated (ASPM) is a centrosomal protein that plays a crucial role in mitotic spindle regulation, neurogenesis, and brain size regulation. Studies reported that ASPM is highly expressed in a variety of cancers and high ASPM expression is related to poor overall survival of LUAD patients46. As a critical mitotic checkpoint gene, cyclin-dependent kinase 1 (CDK1) upregulation may be indicative of poor survival and higher risk for cancer recurrence. CDK1 could be a potential prognostic marker gene in LUAD patients47. Spindle pole body component 25 (SPC25) acts as a key component of the kinetochore associated NDC80 complex48, which is required for chromosome segregation and spindle checkpoint activity. SPC25 expression was enhanced in different kinds of malignant tumors, such as liver, endometrial, and lung cancer. The study in Ref. 48 verified that SPC25 was a potential prognostic biomarker for poor overall survival in LUAD patients.
In summary, all these genes have biological functions associated with mitotic cytokinesis and spindle behavior of mitotic cell division. To validate whether these identified 13 hub genes are of prognostic significance, we analyzed the correlation of their expression levels with LUAD patients' survival. As shown in Supplementary Fig. S2, we found that all these 13 hub genes were upregulated expressed and their high expression is correlated with poor survival of LUAD patients. This evidence gives support to the prognostic significance of these hub genes for LUAD patients. In this sense, our proposed method has the benefit of capturing high-order interactions among gene features to make accurate survival predictions.
In conclusion, we proposed a novel CNN-Cox model which is a CNN-based survival prediction model, combining with the effective feature selection to extract prognosis-related genes from gene expression data. Compared with the existing state-of-the-art survival analysis models, our developed CNN-Cox model achieved more robust superior prediction accuracy on various cancer type datasets. In addition, the simplified CNN design based on simpler 1D convolution operations induces the reduction of the training cost, which is highly desirable in genomics studies. This also allows us to perform a model interpretation to elucidate prognosis markers for each cancer type. Despite of our efforts, the overfitting problem remains challenging for gene expression data analysis tasks. In future works, we plan to utilize the alternative transfer learning strategy to improve the survival prediction, by pretraining deep learning models on the source dataset with sufficient samples and fine-tuning survival analysis models on the final target dataset.
Data availability
The TCGA pan-cancer RNA-seq datasets in the current study are available in the UCSC Xena repository (https://xenabrowser.net/datapages/).
Code availability
The codes of this paper are available at https://github.com/wangwangCCChen/CNN-Cox.
References
Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med 375, 1109–1112 (2016)
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas pan-cancer analysis project. Nat Genet 45, 1113–1120 (2013)
Bindal N, Forbes SA, Beare D, Gunasekaran P, Leung K, Chai YK, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Genome Biol 12, 1–25 (2011)
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012)
Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, et al. Meta-analysis of gene expression profifiles in breast cancer: toward a unifified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 10, R65 (2008)
Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, et al. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology 141, 476–485 (2011)
Lee S, Lim H. Review of statistical methods for survival analysis using genomic data. Genomics Inform 17, e41 (2019)
Lynch CM, Abdollahi B, Fuqua JD, De AR, Bartholomai JA, Balgemann RN, et al. Prediction of lung cancer patient survival via supervised machine learning classification techniques. Int J Med Inform 108, 1–8 (2017)
Wang P, Li Y, Reddy CK. Machine learning for survival analysis: a survey. ACM Comput Surv 51, 1–36 (2019).
Cox DR. Regression models and life-tables. J R Stat Soc Series B Stat Methodol 34, 187–202 (1972).
Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw 39, 1–13 (2011)
Binder H, Schumacher M. Allowing for mandatory covariates in boosting estimation of sparse high dimensional survival models. BMC Bioinformatics 9, 14 (2008)
Zupan B, Demšar J, Kattan MW, Beck JR, Bratko I. Machine learning for survival analysis: a case study on recurrence of prostate cancer. Artif Intell Med 20, 59–75 (2000)
Hofner B, Hothorn T, Kneib T. Variable selection and model choice in structured survival models. Comput Stat 28, 1079–1101 (2013)
Chen Y, Jia Z, Mercola D, Xie X. A gradient boosting algorithm for survival analysis via direct optimization of concordance index. Comput Math Methods Med 2013, 873595 (2013)
Ishwaran H, Kogalur UB, Chen X, Minn AJ. Random survival forests for high-dimensional data. Stat Anal Data Min 4, 115–132 (2011)
Khan FM, Zubek VB. Support vector regression for censored data (SVRc): a novel tool for survival analysis. Proc IEEE Int Conf Data Min 863–868 (2008)
Faraggi D, Simon R. A neural network model for survival data. Stat Med 14, 73–82 (1995)
LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 521, 436–444 (2015)
Shin HC, Roth HR, Gao M, Lu L, Xu Z, Nogues I, et al. Deep convolutional neural networks for computer-aided detection: CNN architectures, dataset characteristics and transfer learning. IEEE Trans Med Imaging 35, 1285–1298 (2016)
Tian T, Wan J, Song Q, Wei Z. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat Mach Intell 1, 191–198 (2019)
Hou X, Wang K, Zhong C, Wei Z. St-trader: A spatial-temporal deep neural network for modeling stock market movement. IEEE/CAA J Autom Sinica 8, 1015–1024 (2021)
Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 18, 851–869 (2016)
Ching T, Zhu X, Garmire LX. Cox-nnet: an artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput Biol 14, 1–18 (2018)
Way GP, Greene CS. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. Pac Symp Biocomput 23, 80–91 (2018)
Kim S, Kim K, Choe J, Lee I, Kang J. Improved survival analysis by learning shared genomic information from pan-cancer data. Bioinformatics 36, i389–i398 (2020)
Sharma A, Vans E, Shigemizu D, Boroevich KA, Tsunoda T. DeepInsight: a methodology to transform a non-image data to an image for convolution neural network architecture. Sci Rep 9, 11399 (2019)
Lyu B, Haque A. Deep learning based tumor type classification using gene expression data. Proc 2018 ACM Int Conf on Bioinformatics, Computational Biology and Health Informatics 89–96 (2018)
Ma S, Zhang Z. OmicsMapNet: transforming omics data to take advantage of deep convolutional neural network for discovery. CoRR abs/1804.05283 (2018)
Lopez-Garcia G, Jerez JM, Franco L, Veredas FJ. Transfer learning with convolutional neural networks for cancer survival prediction using gene-expression data. PLoS ONE 15, e0230536 (2020)
Shin B, Park S, Hong JH, An HJ, Chun SH, Kang K, et al. Cascaded Wx: a novel prognosis-related feature selection framework in human lung adenocarcinoma transcriptomes. Front Genet 10, 1–9 (2019)
Goldman M, Craft B, Brooks AN, Zhu J, Haussler D. The ucsc xena platform for cancer genomics data visualization and interpretation. https://doi.org/10.1101/326470 (2018)
Mostavi M, Chiu YC, Huang Y, Chen Y. Convolutional neural network models for cancer type prediction based on gene expression. BMC Med Genomics 13, 44 (2020)
Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res 24, 1248–1259 (2017)
Katzman JL, Shaham U, Cloninger A, Bates J, Jiang T, Kluger Y. DeepSurv: personalized treatment recommender system using a cox proportional hazards deep neural network. BMC Med Res Methodol 18, 1–12 (2018)
Demiar J, Schuurmans D. Statistical comparisons of classifiers over multiple data sets. J Mach Learn Res 7, 1–30 (2006)
Hao J, Kim Y, Mallavarapu T, Oh JH, Kang M. Cox-PASNet: pathway-based sparse deep neural network for survival analysis. IEEE Int Conf Bioinformatics and Biomedicine 381–386 (2018)
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550 (2005)
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015)
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39, 561 (2011)
Tuan NM, Lee CH. Role of Anillin in tumour: from a prognostic biomarker to a novel target. Cancers (Basel) 12, 1600 (2020)
Wang MY, Chen DP, Qi B, Li MY, Zhu YY, Yin WJ, et al. Pseudogene RACGAP1P activates RACGAP1/Rho/ERK signalling axis as a competing endogenous RNA to promote hepatocellular carcinoma early recurrence. Cell Death Dis 10, 426 (2019)
Hou G, Dong C, Dong Z, Liu G, Xu H, Chen L, et al. Upregulate KIF4A enhances proliferation, invasion of hepatocellular carcinoma and indicates poor prognosis across human cancer types. Sci Rep 7, 41–48 (2017)
Kawai Y, Shibata K, Sakata J, Suzuki S, Utsumi F, Niimi K, et al. KIF20A expression as a prognostic indicator and its possible involvement in the proliferation of ovarian clearcell carcinoma cells. Oncol Rep 40, 195–205 (2018)
Zhang L, Zhu G, Wang X, Liao X, Huang R, Huang C, et al. Genomewide investigation of the clinical significance and prospective molecular mechanisms of kinesin family member genes in patients with lung adenocarcinoma. Oncol Rep 42, 1017–1034 (2019)
Chen Y, Jin L, Jiang Z, Liu S, Feng W. Identifying and validating potential biomarkers of early stage lung adenocarcinoma diagnosis and prognosis. Front Oncol 11, 644426 (2021)
Shi YX, Zhu T, Zou T, Zhuo W, Chen YX, Huang MS, et al. Prognostic and predictive values of CDK1 and MAD2L1 in lung adenocarcinoma. Oncotarget 7, 85235–85243 (2016)
Chen J, Chen H, Yang H, Dai H. SPC25 upregulation increases cancer stem cell properties in non-small cell lung adenocarcinoma cells and independently predicts poor survival. Biomed Pharmacother 100, 233–239 (2018)
Acknowledgements
The authors greatly appreciate all the anonymous reviewers and associate editors for their invaluable, detailed, and constructive suggestions for the manuscript.
Funding
This research was supported by grants from Natural Science Basic Research Program of Shaanxi (No. 2022JM-026) and National Natural Science Foundation of China (No. 61872284, No. 12101482, and No. 12001418).
Author information
Authors and Affiliations
Contributions
QY performed study methodology, experiments designs, model interpretation, and drafted the manuscript. WC performed data processing, model construction, algorithm implementation, and results visualization. ZW and CZ provided review and revision of the manuscript. All authors read and approved the final submitted version of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yin, Q., Chen, W., Zhang, C. et al. A convolutional neural network model for survival prediction based on prognosis-related cascaded Wx feature selection. Lab Invest 102, 1064–1074 (2022). https://doi.org/10.1038/s41374-022-00801-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00801-y
This article is cited by
-
Synthesis of Hybrid Data Consisting of Chest Radiographs and Tabular Clinical Records Using Dual Generative Models for COVID-19 Positive Cases
Journal of Imaging Informatics in Medicine (2024)
-
Explainable and visualizable machine learning models to predict biochemical recurrence of prostate cancer
Clinical and Translational Oncology (2024)
-
Genomic and immunogenomic analysis of three prognostic signature genes in LUAD
BMC Bioinformatics (2023)
-
A texture-based method for predicting molecular markers and survival outcome in lower grade glioma
Applied Intelligence (2023)
