
Abstract
The interplay between extracellular matrix (ECM) stiffness and the tumor microenvironment is increasingly recognized as a critical factor in cancer progression and the efficacy of immunotherapy. This review comprehensively discusses the key factors regulating ECM remodeling, including the activation of cancer-associated fibroblasts and the accumulation and crosslinking of ECM proteins. Furthermore, it provides a detailed exploration of how ECM stiffness influences the behaviors of both tumor and immune cells. Significantly, the impact of ECM stiffness on the response to various immunotherapy strategies, such as immune checkpoint blockade, adoptive cell therapy, oncolytic virus therapy, and therapeutic cancer vaccines, is thoroughly examined. The review also addresses the challenges in translating research findings into clinical practice, highlighting the need for more precise biomaterials that accurately mimic the ECM and the development of novel therapeutic strategies. The insights offered aim to guide future research, with the potential to enhance the effectiveness of cancer immunotherapy modalities.
Similar content being viewed by others
Facts
-
1.
Extracellular matrix (ECM) stiffness plays a critical role in promoting cancer initiation and progression by regulating the malignant behaviors of cancer cells.
-
2.
Key factors regulating ECM remodeling include the activation of cancer-associated fibroblasts and the accumulation and crosslinking of ECM proteins.
-
3.
ECM stiffness forms a formidable physical barrier around tumors, hindering immune cell infiltration and impeding the precise delivery of immunotherapeutic agents.
Open questions
-
1.
What is the role of ECM stiffness in altering the tumor microenvironment, and how does it subsequently affect tumor progression and therapeutic outcomes?
-
2.
What are the effects and underlying mechanisms of ECM stiffness on immune cell behaviors within the tumor microenvironment?
-
3.
How can targeting ECM stiffness be optimized to achieve maximal efficacy in various types of cancer immunotherapy?
Introduction
The process of tumorigenesis depends on two primary factors: genetic and epigenetic alterations within tumor cells, and the dynamic interactions within the tumor microenvironment (TME). The immunosuppressive properties of the TME are crucial in dampening the host’s immune response. The TME includes blood vessels, immune cells, stromal cells, and the extracellular matrix (ECM). Notably, the ECM consists of proteins, mainly divided into fibrous proteins and glycosaminoglycans (GAGs), providing both structural and biochemical support to the resident cells [1, 2]. ECM stiffness, a critical biomechanical property, denotes the resistance of the ECM to deformation when subjected to mechanical stress. It is quantitatively assessed in terms of the elastic modulus. In a physiological context, the rigidity of the ECM is finely balanced, integral to maintaining cellular homeostasis and tissue architecture. However, aberrant alterations in ECM stiffness are implicated in many pathological conditions, particularly in cancer. The ECM dynamically adapts to the progression of cancer, exhibiting shifts in its composition, topological structure, directionality, and notably, its mechanical properties, which directly influence ECM stiffness [3]. These alterations in ECM stiffness subsequently drive changes in cellular mechanobiology, which includes modifications in cytoskeletal tension and the activation of mechanotransduction signaling pathways. Such transformations are pivotal in the evolution of tumors, contributing to their progression towards a more malignant state [4].
Immunotherapy, leveraging the host’s immune system to target malignant cells, has significantly transformed cancer treatment. Compared to traditional therapies such as chemotherapy, this approach exhibits fewer off-target effects and has substantially improved outcomes in various advanced malignancies. However, tumor cells utilize various strategies, including increased ECM stiffness, to impair the efficacy of immunotherapy, leading to inadequate clinical responses in some patients. For instance, elevated ECM stiffness forms a formidable physical barrier around tumors, hindering immune cell infiltration and hampering precise delivery of immunotherapeutic agent. Moreover, increased ECM stiffness can amplify cancer’s immune evasion by altering the expression of immune checkpoint (IC) molecules like PD-L1, PD-1, and CTLA-4, potentially resulting in the failure of IC blockade (ICB) therapy.
Although several reviews have addressed the clinical relevance of the ECM in cancer therapy, the distinctive contributions of our review are noteworthy for a variety of reasons [5,6,7]. First, it offers a timely and comprehensive synthesis of the latest research exploring the impact of ECM stiffness on tumor and immune cell behaviors. Second, the review extensively discusses the effects of targeting ECM stiffness to enhance the efficacy of a range of immunotherapeutic strategies, including ICB therapy, adoptive cell therapy (ACT), oncolytic virus therapy (OVT), and therapeutic cancer vaccines (TCVs). This thorough analysis presents a multifaceted perspective on the potential of ECM modulation in cancer treatment. Lastly, it underscores the emerging challenges and introduces innovative considerations for targeting ECM stiffness, offering novel insights into how this approach could be optimized to improve the efficacy of immunotherapy.
The key factors for regulating ECM remodeling
The increased stiffness in tumors primarily arises from ECM remodeling processes. Enzymes such as matrix metalloproteinases (MMPs), the lysyl oxidase (LOX) family, and Procollagen-Lysine,2-Oxoglutarate 5-Dioxygenase (PLOD) family play a pivotal role in this remodeling. Key factors that regulate ECM remodeling include the activation of cancer-associated fibroblasts (CAFs) and the accumulation and crosslinking of ECM proteins (Fig. 1) [1].

The primary contributors to ECM stiffness include the activation of CAFs, excessive deposition of ECM components, and enhanced collagen crosslinking.
Activation of CAFs
Within the TME, fibroblasts frequently undergo a transition into a persistently activated state known as CAFs. This transformation is induced by growth factors secreted by cancer cells, including transforming growth factor-beta (TGF-β), epidermal growth factor, and bone morphogenetic protein. CAFs exhibit a distinct elongated morphology and lack the markers typical of epithelial cells, endothelial cells, and leukocytes. Importantly, they do not carry the genetic mutations characteristic of cancer cells [8]. Central to the synthesis of various ECM components, CAFs also respond to alterations in the ECM’s physical properties by converting mechanical signals into biochemical responses, which subsequently regulate the expression of specific genes and growth factors.
TGF-β plays a crucial role in the transformation of fibroblasts into CAFs, with integrins being essential for TGF-β activation. Notably, the expression of integrin αvβ6 in cancer cells activates TGF-β, which is positively correlated with increased levels of CAF markers such as α-SMA and fibroblast activation protein (FAP) [9]. The knock-out of the integrin α3 subunit can revert CAFs to an inactivated fibroblast state, effectively reducing cancer cell invasion [10]. Beyond TGF-β1, SPIN90 and specific pro-inflammatory cytokines are critical for CAF activation. A decrease in SPIN90 levels leads to increased microtubule acetylation in CAFs, thereby promoting their activation. Furthermore, reduced expression of SPIN90 activates CAFs through the periostin-FAK-ROCK signaling pathway [11]. Additionally, the pro-inflammatory cytokine IL-1α is capable of reprogramming standard fibroblasts into CAFs, which subsequently results in increased matrix stiffness [12].
Excessive deposition of ECM components
As ECM stiffness increases, there is an accumulation of various ECM components, particularly collagen. In cancer cells, the methylation of the RASSF1A promoter triggers a sequence of events where increased expression of nuclear YAP1 and P4HA2 leads to enhanced collagen deposition in the ECM, thereby contributing to its increased stiffness [13]. HSP47, an endoplasmic reticulum molecular chaperone, facilitates procollagen folding and maturation; its upregulation accelerates collagen secretion and deposition [14]. Beyond collagen, other components such as fibronectin (FN), laminin, elastin, and GAGs significantly modulate ECM stiffness. CAFs produce an FN-rich ECM, with factors like obesity further exacerbating FN accumulation and thereby increasing ECM stiffness [15, 16]. Additionally, ROCK (Rho kinase), acting as a mechanosensor for matrix stiffness, augments tissue stiffness by regulating the synthesis of collagen, FN, and laminin through the β-catenin signaling pathway [3]. An increase in GAG content, induced by YAP activation, leads to increased ECM stiffness, whereas a reduction in GAGs decreases stiffness [17, 18]. Hyaluronic acid (HA), prevalent in the ECM of many fibrosis-associated tumors, profoundly influences their biochemical and mechanical properties. Elevated HA levels are linked with vascular collapse, hypoxia, drug resistance, and a more aggressive tumor phenotype. In metastatic colorectal cancer patients, increased HA and sulfated GAGs (sGAG) following anti-VEGF therapy contribute to enhanced ECM stiffness, creating a physical barrier that hinders drug delivery [19].
Protease families, such as MMPs, adamalysins, and meprins, also play a critical role in ECM remodeling [20]. In cancer, proteases not only facilitate tumor growth and metastasis by altering the ECM structure but also release bioactive ECM fragments to reshape the TME [21]. Specifically, the MMP family, with over 20 zinc-dependent endopeptidases [22], is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs). This regulation is vital in balancing ECM degradation and synthesis [23]. MMPs, particularly MMP-9 and MMP-2, are instrumental in cancer-related processes. For instance, MMP-9-mediated degradation of FN alters αvβ6 integrin dynamics, enhancing β6 integrin expression and promoting cancer cell invasion [24]. Similarly, MMP-2 activity, upregulated by TEM1, promotes tumor invasion and metastasis by enhancing ECM adhesion and degradation [25]. Importantly, growth factors released from ECM cleaved by MMPs contribute to tumor development. In colorectal cancer, neutrophil-derived MMP-9 releases VEGF from the ECM by cleaving heparan sulfates, enhancing endothelial cell sprouting and angiogenesis, underlining MMP-9’s critical role in regulating VEGF bioavailability and its impact on tumor vascularization [26].
A disintegrin and metalloproteinases (ADAMs) and ADAMs with thrombospondin motifs (ADAMTS) are key protease families involved in ECM remodeling; ADAMs modulate cell-ECM interactions and signal transduction through the cleavage of cell surface molecules, while ADAMTS specifically target ECM components such as proteoglycans and collagens, directly contributing to ECM structural changes and functional dynamics. For instance, ADAM8 indirectly contributes to the breakdown and restructuring of the ECM by regulating MMP9 expression, orchestrating the dynamic interplay between these crucial components in ECM remodeling [27, 28]. Additionally, other tissue proteases, such as serine, aspartic, and cysteine proteases, also play significant roles in ECM remodeling [29]. For instance, matriptase plays a key role in cancer progression by altering ECM components such as laminin-332, impacting cell motility and epithelial carcinogenesis [30].
Collagen crosslinking
In the multifaceted stages of collagen synthesis, crosslinking plays a crucial role, organizing procollagen chains into a cohesive network of collagen triple helices. This process begins with lysyl hydroxylases, encoded by the PLOD family, which hydroxylate lysine residues. Following this, the LOX family facilitates the oxidative deamination of lysine and hydroxylysine, forming reactive aldehydes. These aldehydes spontaneously form crosslinks with adjacent lysine or hydroxylysine residues, consequently enhancing ECM stiffness.
Hydroxylysine aldehyde-derived collagen cross-links (HLCC) predominate in high-stiffness tissues like bone, whereas lysine aldehyde-derived collagen cross-links (LCC) are more common in low-stiffness tissues such as connective tissue. PLOD2 promotes collagen crosslinking and hydroxylates terminal peptidyl lysine residues in collagen, leading to a stromal shift marked by increased HLCC and reduced LCC [31]. The LOX family comprises five copper-dependent amino oxidases: LOX and the LOX-like (LOXL) proteins LOXL-1, LOXL-2, LOXL-3, and LOXL-4 [32]. Tumor-initiating cells secrete LOX to catalyze collagen crosslinking, which initiates a series of events including the upregulation of integrin α7, FAK/Src phosphorylation, and ERK1/2 phosphorylation, ultimately resulting in increased matrix stiffness. Similarly, LOXL2 is pivotal in collagen crosslinking and central to the deposition of insoluble collagen during ECM stiffening.
The effects of ECM stiffness on tumor cells
The stiffness of the ECM significantly influences the malignant behaviors of cancer cells, including morphological changes, proliferation, metabolic reprogramming, epithelial-mesenchymal transition (EMT), invasion, metastasis, and resistance to chemotherapy (Fig. 2).

Elevated ECM stiffness induces morphological changes in tumor cells, primarily in the form of pseudopodia formation, expanded spreading areas, and enhanced cell adhesion. These changes promote cancer cell proliferation. Additionally, an increase in ECM stiffness drives metabolic reprogramming in cancer cells. Furthermore, EMT facilitated by ECM stiffness contributes to distant metastasis through a sequence of stages. Notably, increased ECM stiffness creates a physical barrier in the TME, which enhances drug efflux, reduces immune cell cytotoxicity, and impedes the infiltration of oxygen, drugs, and immune cells.
The effects of ECM stiffness on morphology and proliferation of cancer cells
Under conditions of increased ECM stiffness, cancer cells adopt a morphology conducive to adhesion, proliferation, and invasion, thereby facilitating cancer progression. For instance, HCC cells display a compact, rounded shape on softer matrices, whereas on stiffer matrices, they transition to a flattened and extensively spread morphology [33]. Similarly, cells on stiff matrices demonstrate increased spreading, notable actin filament assembly, pronounced pseudopodia formation and enhanced focal adhesion formation [34,35,36,37]. Notably, there is a direct correlation between cell morphology and proliferation: expansive cell spreading and pseudopodia extension are crucial for efficient nutrient absorption, leading to increased cell proliferation. Intriguingly, the integrin αV-FAK axis senses ECM stiffness and induces autophagy in stromal cells via AMPKα, supporting the proliferation of adjacent cancer cells [38]. Additionally, increased ECM stiffness activates the FAK-Rho-ERK signaling cascade, leading to MAPK activation. This cascade promotes aberrant proliferation of mammary epithelial cells and heightens susceptibility to breast carcinoma [39].
The effects of ECM stiffness on metabolic reprogramming of cancer cells
Cancer metabolic reprogramming not only provides energy but also generates essential metabolites necessary for biosynthesis, sustained proliferation, and tumorigenesis. Key metabolic pathways, including aerobic glycolysis, glutaminolysis, macromolecule synthesis, and redox homeostasis, are central to the proliferation and progression of cancer cells [40]. A reciprocal relationship exists between ECM stiffness and cancer cell metabolism. The altered metabolic profiles in cancer cells are associated with ECM remodeling, demonstrating the intricate interplay between tumor mechanical properties and cancer metabolic adaptations. Notably, aspartate derived from CAFs supports cancer cell proliferation, while glutamate from cancer cells maintains the redox balance of CAFs, facilitating ECM modification [41]. Additionally, increased ECM stiffness enhances aerobic glycolysis in cancer cells via YAP activation, contributing to cancer cell migration [42, 43].
The effects of ECM stiffness on EMT of cancer cells
EMT is a cellular process where polarized epithelial cells, in contact with collagen IV at the basal surface, transform to acquire a mesenchymal phenotype. This transformation is characterized by increased migratory and invasive capabilities, enhanced resistance to apoptosis, and elevated production of ECM components [44]. Signals originating from ECM stiffness initiate EMT, driving the transformation of cell morphology towards a mesenchymal phenotype and enhancing their invasive and migratory behaviors [45]. Specifically, ECM stiffness modulates cancer cell migration and invasion by promoting the expression of N-cadherin, while concurrently reducing the levels of E-cadherin [46,47,48]. Furthermore, enzymes responsible for ECM remodeling, which often exhibit increased expression during EMT, are prevalent in aggressive cancer phenotypes [49].
The effects of ECM stiffness on invasion and metastasis of cancer cells
The ECM plays an active role in cancer cell invasion and metastasis processes. Cancer cells, through their interaction with the ECM, are better equipped to invade the surrounding stroma. MMPs, particularly MMP-2 and MMP-9, are key in governing cancer invasion and metastasis by degrading the ECM and promoting EMT. Elevated ECM stiffness activates the FAK/RhoA/ROCK and PI3K/AKT signaling pathways via integrins, thereby increasing the expression of MMP2 and MMP9, and enhancing the invasiveness of cancer cells [50]. Importantly, the role of MMP-9 in response to ECM stiffness is notably context-dependent. In cancer, increased ECM stiffness often initiates EMT, closely linked with MMP-9 upregulation, thereby facilitating tumor invasion [51]. In contrast, the same increase in ECM stiffness can lead to reduced MMP-9 activity in liver fibrosis, serving as a regulatory mechanism to limit ECM degradation and maintain fibrotic tissue structure [52]. Interestingly, in hepatocellular carcinoma (HCC) cells, higher matrix stiffness can enhance EMT and increase MMP-9 expression [46], suggesting a divergent role of MMP-9 from liver fibrosis to HCC progression. This highlights the importance of cell-specific responses, tissue-specific environments, and differential signaling pathways in determining MMP-9’s function across various stages and conditions.
Cancer metastasis, involving the spread of cancer cells from the primary tumor to distant tissues and organs, primarily encompasses five stages: invasion, intravasation, circulation, extravasation, and colonization. Colorectal liver metastasis is a prominent example of distant metastasis influenced by ECM stiffening. The presence of CAFs in the TME is more pronounced in patients with colorectal cancer liver metastasis, correlating with the role of the fibrosis marker LTBP2 in collagen synthesis and ECM restructuring [53, 54]. Similarly, TCF21 increases perivascular ECM stiffness in peritumoral cells, creating a metastatic microenvironment conducive to stimulating colorectal cancer liver metastasis [53, 55].
The effects of ECM stiffness on drug resistance of cancer cells
Recurring tumors post-treatment often exhibit drug resistance, characterized by significant changes in tumor cell differentiation, immune infiltration, and ECM composition. The stiffness of the ECM plays a crucial role in determining the responsiveness of cancer cells to chemotherapy. For instance, elevated ECM stiffness enhances the activity of the multidrug resistance protein 1 on cell membranes, increasing its ability to expel drugs from cancer cells and thereby conferring chemotherapy resistance [56]. Furthermore, increased tumor stiffness obstructs the efficient delivery of therapeutic drugs within the tumor, diminishing the efficacy of such treatments [57].
The crucial role of biomechanical forces in regulating immune cell function
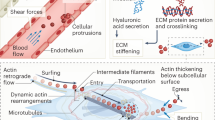
The innate immune arm comprises cells such as macrophages, neutrophils, dendritic cells (DCs), mast cells, basophils, eosinophils, natural killer (NK) cells, and innate lymphoid cells. On the other hand, adaptive immunity primarily involves antigen-specific T cells, with B cells differentiating into plasma cells to produce specific antibodies. Notably, biomechanical forces between immune cells are crucial for maintaining their functions (Fig. 3). T cells are regulated by biomechanical forces that affect their migration, recognition, activation, and effector functions [58]. Furthermore, antigen recognition requires direct physical interactions between T cells and antigen-presenting cells (APCs). During this process, biomechanical forces can modify receptor-ligand bond interactions, affecting the formation of the immunological synapse [59]. LFA1, a key integrin on T and B cells, interacts with adhesion molecules ICAM1 and ICAM2 on APCs, modulating the mechanosensitive antigen recognition process. In addition, B cells selectively internalize high-affinity antigens via contraction mediated by dynamic myosin IIa, influencing the presenting membranes [60]. Similarly, the synapse on CTLs increases the membrane tension of the target cell, enhancing the activity of perforin released by CTLs and amplifying their cytotoxicity [61].

A Force changes detected by immune cell integrins drive movement through retrograde F-actin flow, connected by myosin II. B Membrane tension opens ion channels on immune cells, converting extracellular mechanical cues into intracellular biochemical signals via ion influx. C Under force, immune cells connect to epithelial cells via selectins and roll on their surfaces. At specific sites, immune cell integrins bind to receptors on epithelial cells. Post-adhesion, immune cells develop invadopodia-like protrusions (ILPs), aiding in transmigration. D Antigen recognition necessitates direct contact between T cells and APCs. Mechanical forces influencing receptor-ligand bond formation affect the formation of the immunological synapse. LFA1, the primary integrin on T and B cells, binds tightly to adhesion molecules ICAM1 and ICAM2 on APCs, governing mechanosensitive antigen recognition. E B cells selectively internalize high-affinity antigens via myosin IIa-driven contraction, pulling and invaginating the presenting membranes. F CTLs tighten synapses on target cell membranes by applying force, promoting perforin’s pore-forming activity, and enhancing cytotoxicity.
Cells interact with the ECM, and biomechanical forces generated by ECM directly or indirectly influence immune cell fate and function. Therefore, elucidating the relationship between biomechanical force and ECM stiffness is essential in determining cellular responses. In this dynamic, the biomechanical force is primarily a consequence of the ECM’s stiffness, exerted upon the cells [62]. A stiffer ECM provides greater mechanical resistance, which cells sense and respond to. This interaction is critical for mechanotransduction, where cells interpret these mechanical cues from the ECM and translate them into biochemical signals [63]. This process significantly influences cellular behaviors. For instance, the biomechanical force derived from stiff lymph nodes, sensed by the mechanosensor YAP, suppresses T cell proliferation by inhibiting NFAT1’s nuclear translocation [64].
The effects of ECM stiffness on immune cells
The stiffness of ECM profoundly influences the activation, polarization, migration, infiltration, antigen presentation, and cytotoxicity of immune cells, ultimately affecting the efficacy of cancer immunotherapy. Stiffer tumors often display decreased immune cell density at their core compared to softer ones. The migration of immune cells within the ECM is governed by the interplay and spatial arrangement of its constituents, with collagen fibers playing a critical role [65]. Elevated ECM stiffness and dense collagen networks can impede immune cell infiltration and motility, thereby diminishing the cytotoxic interactions between immune and cancer cells [66].
The effects of CAFs on immune cells
It is well documented that CAFs play a pivotal role in modulating ECM stiffness and impacting immune cell function [67]. They contribute to immune evasion by creating a physical barrier that impedes immune cell infiltration and by secreting a range of cytokines and chemokines that can alter immune cell recruitment, activation, and function [6]. For instance, CAFs in esophageal cancer negatively correlate with CD8+ TILs and positively with FoxP3+ TILs, indicating a role in tumor immunosuppression. Elevated IL6 secretion in CAF-cancer cell cocultures leads to altered TIL profiles, with IL6 blockade reversing these effects [68]. Similarly, CAFs directly suppress cytotoxic T cells, using mechanisms like antigen processing, cross-presentation, and PD-L2 and FASL-mediated killing. This suppression is evident in human tumors and can be counteracted in vitro and in vivo by neutralizing PD-L2 or FASL [69]. Notably, CD70 expression on a specific subset of CAFs in invasive CRC serves as an independent adverse prognostic marker. These CD70-positive CAFs facilitate tumor migration and increase Treg frequency, contributing to immune escape in CRC and underscoring the potential of CD70-targeting antibodies in therapy [70]. In oral squamous cell carcinoma, the CD68+ CAFs vary in distribution within the tumor and are involved in immune modulation, notably by enhancing Treg recruitment through chemokines CCL17 and CCL22 [71].
In addition to T cells, CAFs also exert significant influence on other immune cells, such as neutrophils, macrophages, and NK cells, impacting various aspects of the immune response within the TME. CAFs influence neutrophil chemotaxis, survival, and activation in HCC mediated by an IL6-STAT3-PDL1 signaling pathway [72]. This interaction impairs T-cell function, underscoring a novel mechanism of CAFs in tumor microenvironment modulation and identifying a potential therapeutic target in HCC. In breast cancer, CAFs drive monocyte recruitment and polarization towards immunosuppressive M2-like macrophages, enhancing cancer progression and shaping an immunosuppressive microenvironment [73]. Interestingly, in colorectal cancer, CAFs promote monocyte adhesion and M2 macrophage polarization through IL-8 secretion and VCAM-1 upregulation, working synergistically with TAMs to suppress NK cell function [74]. In addition, CAFs in HCC induce NK cell dysfunction, characterized by reduced cytotoxicity and impaired cytokine production, through PGE2 and IDO [75]. Importantly, in esophageal squamous cell carcinoma, CAFs facilitate the differentiation of monocytes into M-MDSCs, enhancing drug resistance via IL-6 and miR-21-mediated STAT3 activation [76]. This interaction between CAFs and M-MDSCs correlates with poorer patient survival, highlighting a potential target in STAT3 signaling for overcoming chemotherapy resistance.
The effects of ECM stiffness on T cell functionalities
T cells recognize peptide antigens derived from intracellular protein degradation and are classified into two main subsets based on surface glycoproteins: CD4+ and CD8+ T cells. CD8+ T cells, also known as CTLs, directly target and eliminate virus-infected and cancerous cells. CD4+ T cells, often referred to as “helper T cells,” assist in amplifying the immune response by activating memory B cells and CD8+ T cells. The adaptive immune response’s efficacy in preventing tumor formation relies on the ability of CTLs to identify foreign or mutated antigens on transformed cells and subsequently induce cell death via T-cell-mediated cytotoxicity.
Increased ECM stiffness can impair T cell migration and infiltration by forming barriers through ECM remodeling. For instance, loosely arranged collagen fibers within the ECM create a conducive environment for immune cell migration, facilitating efficient movement of T and B lymphocytes [77]. In addition, attributes of collagen fibers, such as orientation and density, influence the migration patterns of resident CD8+ T cells within tumors [78]. Elevated ECM stiffness leads to increased tenascin-C expression, which acts as a physical barrier, hindering T-cell movement and infiltration [79,80,81]. Moreover, the fibrotic ECM directly impacts T cells, resulting in limited infiltration in solid tumors and a predominantly immunosuppressive TME [82, 83]. Interestingly, βig-h3, predominantly produced in the ECM by CAFs, can be targeted to reduce tumor growth by enhancing the numbers of activated CTLs [84]. Notably, SOD3, a crucial modulator of tumor laminin, facilitates the selective infiltration of T lymphocytes into tumors, transforming inactive tumors into immunologically active ones [85].
In addition to migration and infiltration, ECM stiffness critically regulates T cell including proliferation, differentiation, and cytotoxic function. For example, a less stiff matrix enhances the proliferation of both CD4+ and CD8+ T cells and increases IL-2 secretion [86, 87]. In addition, signaled through the mechanosensor Piezo1, ECM stiffness influences the differentiation balance between Th1 cells and regulatory T cells (Tregs) [88]. Moreover, for an effective immune response, T cells need to increase nutrient uptake. However, T cells can be deprived of essential nutrients due to ECM stiffness resulting from vascular calcification and metabolic shifts, thereby impeding effector T cell differentiation and function [89]. Importantly, High collagen concentration within tumors correlates with fewer infiltrating CD8+ T cells and a higher CD4+/CD8+ ratio, along with reduced expression of cytotoxic activity markers [86, 90]. Similarly, CTL-based therapies are less effective in collagen-rich environments as ECM density and porosity significantly impact CTL performance [91]. Interestingly, microtubule inhibitors like colchicine can improve the cytotoxic efficiency of CTLs in collagen-rich environments [91]. Notably, the use of TGF-β blockade antibodies in combination with anti-PD-L1 antibodies reduces TGF-β signaling in stromal cells, thereby enhancing T cell infiltration and triggering potent anti-tumor immune responses [92].
The effects of ECM stiffness on tumor-associated macrophages
Macrophages, originating from monocytes in the bone marrow, enter the bloodstream as monocytic precursors and differentiate into fully mature macrophages upon infiltrating tissues and organs. These cells can be categorized into two distinct phenotypes: “classically activated macrophages” or M1, exhibiting a pro-inflammatory phenotype, and “alternatively activated macrophages” or M2, known for their anti-inflammatory characteristics. M1-like macrophages release molecules such as reactive oxygen intermediates, reactive nitrogen intermediates, and TNF-α to suppress tumor growth. In contrast, M2-like macrophages facilitate tumor progression and metastasis through the secretion of matrix-degrading enzymes, vascular growth factors, immunosuppressive cytokines, and chemotactic factors.
ECM stiffness significantly influences the morphology, cytoskeletal arrangement, functionality, and metabolism of macrophages. On high-stiffness substrates, macrophages adopt an elongated, spindle-like shape, with an extended spreading area and increased formation of filamentous pseudopodia. Elevated substrate stiffness also enhances the expression of CSF-1 in cancer cells, driving macrophages towards an M2-like phenotype. Conversely, pliable substrates induce reactive oxygen species (ROS) production in macrophages, leading to polarization to the M1 phenotype via the ROS-mediated NF-κB pathway, and resulting in increased secretion of cytokines IL-1β and TNF-α [93, 94]. Additionally, interactions between βig-h3 and type I collagen form denser fibers. Macrophages cultured on these fibers undergo morphological changes, exhibiting an enhanced M2 phenotype and amplified immunosuppressive capabilities [95].
The effects of ECM stiffness on diverse immune cell function
The implications of ECM stiffness also extend to various immune cell types. For example, the protein STEAP3, whose activity is influenced by matrix stiffness, facilitates neutrophil infiltration [96]. Upon activation, neutrophils can release chromatin to form neutrophil extracellular traps as part of their immune defense strategy. However, increased matrix stiffness may impair this response [97]. Additionally, SOX9, by increasing collagen deposition and thereby intensifying ECM stiffness, leads to reduced infiltration of DCs within tumors, as well as decreased infiltration of NK and CTL cells [98]. Notably, matrix stiffness influences immature DCs by regulating the expression of β2 integrin and C-type lectin, which affect antigen uptake and podosome formation. It also modulates the expression of CD83 and CCR7 in mature DCs, directing chemokine-driven migration (Fig. 4) [99].

In soft ECM environments, there is enhanced T cell proliferation and guidance of CD8+ T cell activation, migration, and infiltration. Conversely, a stiff ECM hinders T cell proliferation and reduces CD8+ T cell mobility and cytotoxicity. Low ECM stiffness steers naive CD4+ T cells toward Th1 polarization and promotes M1 macrophage polarization from M0, aiding in anticancer immune effects. High ECM stiffness directs immature CD4+ T cells to Th2 polarization and M2 macrophage polarization from M0, fostering pro-cancer TME effects.
The effects of ECM stiffness on tumor endothelial cells (TECs) and pericytes
Tumor progression is closely linked with angiogenesis, characterized by the presence of TECs lining the blood vessels, pericytes enveloping the microvasculature, and smooth muscle cells within arterioles and venules. ECM stiffness plays a crucial role in capillary morphogenesis, network development, and barrier integrity, directly affecting the nature and extent of angiogenesis. This active restructuring of the ECM fosters a perivascular niche favorable for cancer metastasis.
TECs, known for their enhanced proliferative, immune-regulatory, and angiogenic capabilities compared to standard endothelial cells, exhibit energy due to their unresponsiveness to pro-inflammatory stimuli. They hinder the adhesion and migration of immune cells, presenting challenges for immune-based cancer therapies. Persistent abnormal physical signals within the TME disrupt the balance between cellular and ECM forces, influencing the gene expression and behavior of TECs. Additionally, tumors stimulate TECs through the secretion of VEGF, which is further enhanced by ECM stiffening. This increased VEGF secretion enhances TEC proliferation and migration. The YAP activity in TECs, influenced by VEGF and mediated by ECM stiffness, promotes endothelial cell proliferation, migration, and sprouting, driving neovascularization [100]. Notably, TECs are highly responsive to changes in ECM stiffness; enhanced traction forces lead to increased cellular contractility, affecting aberrant angiogenesis [101, 102].
Pericytes, integral components of the vascular wall, are embedded within microvascular collagen IV. Dysfunctional interactions between pericytes and TECs can lead to the formation of disorganized, permeable, and ineffective vascular structures within tumors. During ECM fibrosis, increased matrix stiffness may trigger the conversion of pericytes into fibroblasts. This conversion enhances cancer cell mobility and invasiveness through YAP activation [103, 104]. Interestingly, tumor pericytes contribute to perivascular ECM stiffness. They rearrange collagen and degrade collagen IV, influencing ECM remodeling. This remodeling establishes a perivascular environment that is conducive to cancer metastasis [53].
Targeting ECM stiffness for improving the efficacy of immunotherapy
The clinical achievements of immunotherapy have been both promising and limited. While significant improvements following immunotherapeutic treatments have been observed in a proportion of patients with various cancers, the majority have seen minimal or no benefit from these interventions. The varied responses to immunotherapies are influenced by factors such as tumor heterogeneity, the nature and phase of cancer, the patient’s treatment history, tumor-induced immune suppression mechanisms, and the need to identify specific oncogenic biomarkers and pathways before treatment. Challenges like immune-related side effects, off-target consequences, and immune evasion also persist with immunotherapies.
Single-agent immunotherapy approaches face the challenge of multiple immune resistance mechanisms. This highlights the potential advantage of combination therapies in preventing the emergence of immune-resistant strains. Increased tumor stiffness can impede the effective delivery of immunotherapeutic agents to tumor sites. Recent studies suggest that targeting the tumor ECM with immunotherapies could be particularly effective against treatment-resistant tumors with abundant stroma.
Targeting ECM stiffness for improving the efficacy of ICB therapy
ICB therapy targets T cell regulatory pathways to boost anti-tumor responses. Normally, immune checkpoints on immune cells prevent excessive activation, protecting healthy tissue. In tumors, however, increased checkpoint expression impairs T-cell activity, weakening the immune response against cancer [7]. Notably, the efficacy of ICB therapy is challenged by the dense ECM in tumors, which can hinder immune cell infiltration and limit therapeutic impact.
ECM stiffness has been shown to critically regulate the expression of IC molecules. For instance, an increase in collagen fiber density, leading to enhanced matrix stiffness, may stimulate PD-L1 expression, thereby promoting immune evasion in cancers [105, 106]. Additionally, a deficiency in SNF5 is associated with an increased presence of tumor-infiltrating CD8+ T cells and a reduction in PD-L1-positive cells in vivo. Conversely, increased ECM stiffness can promote cancer cell immune evasion by promoting the activation of the STAT-3 pathway, a consequence of elevated SNF5 expression [107]. Similarly, increased matrix stiffness induces the formation of actin stress fibers, resulting in increased PD-L1 expression in cancer cells. This upregulation can be mitigated using actin polymerization inhibitors [108]. Notably, increased stiffness in the ECM can lead to hypoxia. Under such conditions, HIF-1α, a central transcriptional regulator in the hypoxic response, upregulates PD-L1. Combining HIF-1α inhibitors with anti-PD-L1 antibodies has demonstrated a stronger inhibitory effect on tumor growth [109].
PD-1 is widely expressed on a variety of immune cells, and ECM stiffness is closely associated with PD-1-based immunotherapy. For instance, ECM stiffness can act as an indicator of patient outcomes following treatment with PD-1 inhibitors in combination with Lenvatinib [110]. Additionally, reducing ECM stiffness by targeting collagen crosslinking can enhance T cell migration, thereby improving the efficacy of anti-PD-1 therapies [79]. Moreover, neutrophil elastase, known for its ability to degrade the tumor ECM, facilitates CTL infiltration into tumors and enhances the efficacy of PD-1-based immunotherapy [111]. Interestingly, targeting LOX family members with specific inhibitors can also improve T cell migration, boosting the effectiveness of anti-PD-1 therapies [79]. Similarly, exposing macrophages to LOXL4 can induce an immunosuppressive phenotype and upregulate PD-L1 expression. The synergistic use of ICB and LOXL4 inhibitors presents a promising approach to enhance immunotherapeutic outcomes. Notably, a semiconducting polymer-based agent that reduces ECM stiffness by inhibiting LOX enzyme activity can be combined with immunogenic phototherapy and ICB immunotherapy. This combination augments cytotoxic T cell penetration into tumors, inhibiting tumor proliferation and spread [112]. Furthermore, the simultaneous inhibition of MEK and STAT3 can reprogram CAFs and the immune microenvironment, offering a potential approach to overcome tumor immune resistance to ICIs [113].
CTLA-4 functions by binding to CD80/86 ligands on CD28, subsequently interacting with the TCR. This interaction leads to the cessation of T cell activation. Initial concentrations of certain collagen proteins in the ECM have been linked to response rates and survival outcomes in patients with metastatic melanoma receiving anti-CTLA-4 therapy [114]. Moreover, MMP and granzyme B-mediated degradation of type III and IV collagens may enhance prognoses [115]. Similarly, small molecule inhibitors targeting MMP2/9 can increase antitumor immunity, reduce tumor burden, and extend patient survival. These inhibitors also effectively reduce PD-L1 expression in cancer cells, thereby enhancing the effectiveness of PD-1 or CTLA-4 therapies (Fig. 5A) [116].

A Mechanisms and strategies to counter high ECM stiffness in ICB. Elevated ECM stiffness modulates the expression of ICs through mechanotransduction signaling pathways. ICIs delivery is enhanced when combined with ECM remodeling agents like MMP and LOX inhibitors, enhancing ICB therapeutic efficacy. B Physical factors impeding CAR-T cell delivery and corresponding strategies. In tumors with high ECM stiffness, CAF encapsulation and abnormal vasculature obstruct CAR-T cell infiltration and lead to T-cell exhaustion. Targeting specific proteins, such as EDA, EDB, or CAF marker FAP, facilitates CAR-T cell penetration, effectively curtailing tumor growth. C Physical barriers to OV delivery and strategies to overcome them. The stiff ECM barrier hinders OVs from reaching tumor cells. Pairing OVs with ECM-degrading proteins, such as MMPs, hyaluronidase, or relaxin, breaks down this barrier, amplifying the OV’s cytotoxic effects by T-cell activation and self-replicating. D Therapeutic strategies for overcoming high ECM stiffness impeding TCV delivery. The stiff ECM barrier hampers TCV effects. TCVs combined with ECM-degrading agents promote antigen presentation of DCs and stimulates the activation of CD8+ T cell, thereby intensifying their cytotoxicity against cancer cells.
Targeting ECM stiffness for improving the efficacy of ACT
ACT is a personalized cancer treatment involving the infusion of autologous or allogeneic T cells [117]. Clinical trials have demonstrated significant, sometimes long-lasting, tumor regressions using expanded tumor-infiltrating lymphocytes [118]. The process includes isolating T cells from the patient, engineering them to express chimeric antigen receptors (CARs) or TCRs, cultivating them, and reintroducing them to attack cancer cells [119]. Despite its successes, ACT faces challenges such as antigen escape, effects on normal tissues, limited T cell infiltration into tumors, the suppressive TME, and side effects from modified T cells [120].
The dense structure of the ECM acts as a barrier to CAR-T cell infiltration, due in part to mechanical activation of the TGF-β pathway and abnormal vascular networks [82]. Consequently, CAR-T cells often fail to reach their intended tumor targets, undergo metabolic alterations, and shift towards an immunosuppressive environment, resulting in T cell exhaustion [83]. An innovative approach targeting laminin within the ECM has been shown to enhance immune cell infiltration by degrading the ECM, improving tumor blood flow, and boosting the delivery efficacy of therapeutic agents [121]. Importantly, applications of FN-targeted T cell therapies have shown potential, demonstrating both tolerance and efficacy in advanced cancer cases [122]. Drugs like nattokinase show promise; their administration not only degrades FN but also combats fibrosis enhanced by CAFs, thereby reducing tumor stiffness, enhancing CAR-T cell infiltration, and bolstering the overall efficacy of CAR-T therapy for solid tumors [123]. Specific isoforms of FN, arising from selective mRNA splicing and the addition of structural domains, can influence the outcomes of CAR-T therapy. The extra domain A (EDA) and extra domain B (EDB) are primary FN domains utilized in CAR-T treatments. CAR-T cells targeting EDA have demonstrated anti-angiogenic properties and notable reductions in genes associated with pathways critical to tumor progression, such as EMT, collagen synthesis, IL-6/STAT5, and KRAS pathways [124]. Notably, gold nanoparticles equipped with EDB can induce CTL activation, offering a strategic advantage in cancer immunotherapy [125]. Similarly, CAR-T cell strategies focusing on EDB have shown efficacy in slowing tumor growth through enhanced immune infiltration and promoting tumor cell death [126].
Targeting CAF markers such as FAP with chimeric CAR-T cells has shown promise in limiting tumor growth and enhancing host immunity, while presenting minimal side effects [127]. The introduction of FAP-targeted CAR-T cells has been observed to reduce tumor blood supply and halt tumor growth [128]. In addition, transferring CAR-T cells that target FAP can substantially diminish FAP-expressing stromal cells, leading to inhibition of tumor growth [129]. Furthermore, combining anti-FAP CAR-T cell approaches with PD-1 inhibitors could offer a more effective strategy against tumor progression (Fig. 5B) [130].
Targeting ECM stiffness for improving the efficacy of OVT
Oncolytic viruses (OVs) target and disintegrate cancer cells through oncolysis, simultaneously activating the host’s immune system for an antitumor response [131]. Importantly, OVs possess a natural preference for cancer cells, or can be genetically modified to target specific tumor markers. Once inside the tumor, OVs attach to cell receptors, using the host cell’s machinery to replicate. The resulting breakdown of infected cells releases new viral particles, extending their anti-tumor action [132]. The clinical application of OVT is limited by the rapid development of neutralizing antibodies and potential risks for immunocompromised patients due to intense immune reactions [133, 134]. Notably, the ECM also poses a barrier to deep tumor tissue penetration of OVs [6].
Current combined OVT approaches with ECM-degrading agents include hyaluronidase, core protein polysaccharides, and relaxin. VCN-01, a specially engineered oncolytic adenovirus, replicates and produces hyaluronidase in cancer cells through the disrupted RB1 pathway. The hyaluronidase released by VCN-01 degrades the tumor stroma, reducing tumor stiffness and facilitating the delivery of various therapeutic agents, including chemotherapy and therapeutic antibodies [135]. Similarly, the oncolytic adenovirus ICOVIR17 promotes hyaluronan degradation in the ECM and increases PD-L1 expression in cancer cells and macrophages, enhancing the infiltration of CD8+ T cells and macrophages into the tumor [136]. Core protein polysaccharides, known for degrading ECM components by reducing collagen fiber diameter and modulating TGF-β, also enhance MMP-1 activity. Oncolytic adenoviruses expressing core protein polysaccharides can promote viral spread and elevate their cytotoxic effects against cancer cells [137]. Relaxin, an ECM-degrading protein, improves viral transduction and propagation efficiency, broadens virus distribution and penetration, inhibits tumor growth and metastasis, and improves survival rates post-treatment (Fig. 5C) [138].
Targeting ECM stiffness for improving the efficacy of TCVs
TCVs, which utilize tumor-associated antigens and tumor-specific antigens, stimulate the immune response against cancer, potentially extending remission and improving survival rates. Currently, TCVs are mainly employed alongside traditional therapies for treating small, residual post-surgery tumors, with their broader application constrained by the evolving nature of cancer cells and the immunosuppressive TME [139]. Furthermore, the ECM presents an additional obstacle, acting as a physical barrier to vaccine delivery. Modulating the ECM could improve vaccine infiltration and reshape the TME, potentially enhancing the effectiveness of TCV therapies.
Vaccines targeting ECM stiffness focus on degrading the ECM to boost T lymphocyte activity and cytotoxicity. For instance, TCVs containing the Spam1 gene express hyaluronidase, degrading the ECM, enhancing blood flow, reducing tumor hypoxia, and modulating the immune environment, thereby elevating antitumor efficiency and reducing tumor recurrence risk [140]. In addition, TCVs targeting the EDA domain of FN have shown potential in curbing metastatic tumor progression [141], while those targeting the EDB of FN effectively diminish the bulk of solid tumors [142,143,144]. Moreover, an orally administered DNA vaccine targeting FAP-depleted CAFs decreases type I collagen content, restricting the growth and metastasis of primary tumor cells promoting multidrug resistance in mice [145]. Combined with chemotherapy, this vaccine amplifies anti-tumor effects by increasing chemotherapy drug uptake and accumulation [145, 146]. Likewise, clinical research on a FAP-targeted cancer vaccine has shown promising safety profiles and tumor engagement, enhancing CD8+ T cell-mediated antitumor immunity [147]. Notably, vaccines expressing immature laminin receptor proteins are linked to sustained protective responses mediated by CD8+ T cells [148]. Applying elastin-like polypeptides directly to tumors can impede the growth of specific breast cancers and reduce the incidence of lung metastasis (Fig. 5D) [149].
Enhancing the efficacy of immunotherapy with nanocarriers targeting and degrading the ECM
In addition to the previously mentioned combination therapies, nanocarriers have shown great potential in enhancing cancer immunotherapy outcomes. For example, nanoparticles combined with MMP-2 inhibitors have been reported to improve outcomes in cancer photodynamic immunotherapy [150]. Similarly, MMP-sensitive nanomaterials, designed to degrade at a controlled rate within the TME, facilitate the release of their therapeutic contents and enhance T cell presence at the tumor site [151]. Notably, nanovaccines incorporating laminin peptides have demonstrated efficacy in boosting peptide-specific CD8+ T cell responses, eliciting protective immune reactions with Th1 polarization [152]. Additionally, a HA-based nanovaccine mitigates the immunosuppressive TME by promoting robust infiltration of both CD4+ and CD8+ T cells [153]. Increased ECM stiffness is correlated with elevated levels of sGAGs, such as heparan sulfate and chondroitin sulfate, which align with an intensified immune response to these GAGs [154]. Prodrug nanoparticles using chondroitin sulfate as a base target the Golgi apparatus in tumor cells. This targeting helps to reduce immunosuppression in photodynamic immunotherapy by inhibiting the secretion of immunosuppressive cytokines [155].
Challenges and future prospects
The pivotal role of ECM stiffness in the success and prognosis of cancer immunotherapy, as well as in the precision of delivering immunotherapeutic drugs, is increasingly recognized. Modulating ECM stiffness can significantly influence the efficacy of various immunotherapy strategies, yet this integrative approach also poses challenges in transitioning from research to clinical practice.
Firstly, accurately assessing tumor stiffness is critical for evaluating patient prognosis and responsiveness to immunotherapy. Clinicians employ a range of diagnostic techniques, including manual palpation, color Doppler ultrasound, magnetic resonance elastography, shear wave elastography, and transient elastography, to gauge soft tissue stiffness. The selection of these methods often depends on the specific type of tissue being examined. An integrated approach to these techniques is vital for a comprehensive assessment of tumor stiffness. The stiffness of the ECM, predominantly determined by collagen content and cross-linking, can be indirectly measured through tools like mass spectrometry and Sirius Red staining [3]. However, further research is essential to identify the most accurate and non-invasive methods for measuring tumor stiffness, particularly for early detection.
Secondly, there is an urgent need to develop more inhibitors targeting mechanotransduction signaling pathways or ECM-degrading agents to optimize the co-delivery of immunotherapeutic drugs. Bintrafusp Alfa (M7824), which targets PD-L1 and the mechanotransducer TGF-β, is currently under clinical investigation. Although initial trials have been promising, comprehensive comparisons with standard immunotherapies are crucial [156,157,158,159,160,161,162,163,164]. The exploration of other immunotherapy drugs targeting the ECM is in its nascent stages, underscoring the need for extensive clinical trials to evaluate the safety and efficacy of these potential inhibitors (Table 1).
Thirdly, it is imperative to devise strategies that minimize the side effects of inhibitors targeting mechanotransduction pathways. Given the essential roles of molecules like integrins, TGF-β, and actin in maintaining tissue homeostasis, their inhibition could lead to unintended physiological consequences. Enhancing the specificity of these drugs is critical. One potential approach involves the use of nanoparticles for targeted drug delivery specifically to stiff tumor regions, thereby sparing the surrounding healthy tissues. Additionally, exploring binding sites on challenging proteins such as Rho could broaden the spectrum of combinational treatments with immunotherapy.
Fourthly, an innovative direction in cancer therapy involves targeting CAFs. Transforming CAFs into anti-tumor fibroblasts is crucial for maintaining the mechanical balance of the ECM. Contemporary strategies encompass TCVs targeting the CAF-activating protein FAP, CAR-T cell therapies, oncolytic viruses targeting CAFs, inhibiting cancer cell mechanotransduction to reduce TGF-β secretion, and focusing on pathways such as CXCL12-CXCR4, JAK-STAT3, and Hedgehog.
Lastly, a significant gap in current research is the lack of biomaterials that accurately mimic the complex composition of the ECM. Studies often rely on hydrogels derived from single ECM components, which may not faithfully represent the conditions in human tumors. While natural ECM hydrogels, often sourced from murine tumor collagen IV, provide a closer approximation to in vivo conditions, their dissimilarity to human tumor environments limits their applicability. Consequently, the development of more precise biomaterials and effective animal models is paramount for advancing the field of cancer research and therapy.
Conclusions
In conclusion, the critical role of ECM stiffness in the realm of immunotherapy is increasingly apparent. Dysregulation of ECM stiffness profoundly influences cancer initiation and progression, underscoring its significance in oncological research and treatment. Interdisciplinary collaborations, bridging oncology, cell biology, physics, engineering, materials science, and nanotechnology, are pivotal in advancing therapeutic strategies that target ECM stiffness and mechanotransduction signaling pathways. These collaborative efforts are instrumental in transitioning such strategies from bench to bedside, potentially expanding the scope and enhancing the efficacy of immunotherapy in cancer treatment.
Data availability
The data generated are included within the manuscript.
References
Deng B, Zhao Z, Kong W, Han C, Shen X, Zhou C. Biological role of matrix stiffness in tumor growth and treatment. J Transl Med. 2022;20:540.
Sun B. The mechanics of fibrillar collagen extracellular matrix. Cell Rep Phys Sci. 2021;2:100515.
Jiang Y, Zhang H, Wang J, Liu Y, Luo T, Hua H. Targeting extracellular matrix stiffness and mechanotransducers to improve cancer therapy. J Hematol Oncol. 2022;15:34.
Chin L, Xia Y, Discher DE, Janmey PA. Mechanotransduction in cancer. Curr Opin Chem Eng. 2016;11:77–84.
He X, Lee B, Jiang Y. Extracellular matrix in cancer progression and therapy. Med Rev. 2022;2:125–39.
Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2020;6:160.
Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021;6:153.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–86.
Peng C, Zou X, Xia W, Gao H, Li Z, Liu N, et al. Integrin alphavbeta6 plays a bi-directional regulation role between colon cancer cells and cancer-associated fibroblasts. Biosci Rep. 2018;38:BSR20180243.
Jang I, Beningo K. Integrins, CAFs and mechanical forces in the progression of cancer. Cancers. 2019;11:721.
You E, Huh YH, Kwon A, Kim SH, Chae IH, Lee OJ, et al. SPIN90 depletion and microtubule acetylation mediate stromal fibroblast activation in breast cancer progression. Cancer Res. 2017;77:4710–22.
Enukashvily NI, Ponomartsev NV, Ketkar A, Suezov R, Chubar AV, Prjibelski AD, et al. Pericentromeric satellite lncRNAs are induced in cancer-associated fibroblasts and regulate their functions in lung tumorigenesis. Cell Death Dis. 2023;14:19.
Pankova D, Jiang Y, Chatzifrangkeskou M, Vendrell I, Buzzelli J, Ryan A, et al. RASSF1A controls tissue stiffness and cancer stem-like cells in lung adenocarcinoma. EMBO J. 2019;38:e100532.
Ishikawa Y, Rubin K, Bächinger HP, Kalamajski S. The endoplasmic reticulum-resident collagen chaperone Hsp47 interacts with and promotes the secretion of decorin, fibromodulin, and lumican. J Biol Chem. 2018;293:13707–16.
Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799–816.
Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7:301ra130.
Ghasemi H, Mousavibahar SH, Hashemnia M, Karimi J, Khodadadi I, Tavilani H. Transitional cell carcinoma matrix stiffness regulates the osteopontin and YAP expression in recurrent patients. Mol Biol Rep. 2021;48:4253–62.
Roth J, Hoop CL, Williams JK, Hayes R, Baum J. Probing the effect of glycosaminoglycan depletion on integrin interactions with collagen I fibrils in the native extracellular matrix environment. Protein Sci. 2023;32:e4508.
Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8:360ra135.
Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801.
Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74.
Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13:649–65.
Li W, Liu Z, Zhao C, Zhai L. Binding of MMP-9-degraded fibronectin to beta6 integrin promotes invasion via the FAK-Src-related Erk1/2 and PI3K/Akt/Smad-1/5/8 pathways in breast cancer. Oncol Rep. 2015;34:1345–52.
Wu C, Sun W, Shen D, Li H, Tong X, Guo Y. TEM1 up-regulates MMP-2 and promotes ECM remodeling for facilitating invasion and migration of uterine sarcoma. Discov Oncol. 2023;14:5.
Hawinkels LJ, Zuidwijk K, Verspaget HW, de Jonge-Muller ES, van Duijn W, Ferreira V, et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur J Cancer. 2008;44:1904–13.
Mierke CT. The versatile roles of ADAM8 in cancer cell migration, mechanics, and extracellular matrix remodeling. Front Cell Dev Biol. 2023;11:1130823.
Duan B, Liu Y, Hu H, Shi FG, Liu YL, Xue H, et al. Notch1-ADAM8 positive feed-back loop regulates the degradation of chondrogenic extracellular matrix and osteoarthritis progression. Cell Commun Signal. 2019;17:134.
Fonović M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. 2014;1840:2560–70.
Tripathi M, Potdar AA, Yamashita H, Weidow B, Cummings PT, Kirchhofer D, et al. Laminin-332 cleavage by matriptase alters motility parameters of prostate cancer cells. Prostate. 2011;71:184–96.
Chen Y, Terajima M, Yang Y, Sun L, Ahn YH, Pankova D, et al. Lysyl hydroxylase 2 induces a collagen cross-link switch in tumor stroma. J Clin Investig. 2015;125:1147–62.
Tenti P, Vannucci L. Lysyl oxidases: linking structures and immunity in the tumor microenvironment. Cancer Immunol Immunother. 2020;69:223–35.
Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–205.
Liu B, Kilpatrick JI, Lukasz B, Jarvis SP, McDonnell F, Wallace DM, et al. Increased substrate stiffness elicits a myofibroblastic phenotype in human lamina cribrosa cells. Investig Ophthalmol Vis Sci. 2018;59:803–14.
McKenzie AJ, Hicks SR, Svec KV, Naughton H, Edmunds ZL, Howe AK. The mechanical microenvironment regulates ovarian cancer cell morphology, migration, and spheroid disaggregation. Sci Rep. 2018;8:7228.
Piao J, You K, Guo Y, Zhang Y, Li Z, Geng L. Substrate stiffness affects epithelial-mesenchymal transition of cervical cancer cells through miR-106b and its target protein DAB2. Int J Oncol. 2017;50:2033–42.
Prauzner-Bechcicki S, Raczkowska J, Madej E, Pabijan J, Lukes J, Sepitka J, et al. PDMS substrate stiffness affects the morphology and growth profiles of cancerous prostate and melanoma cells. J Mech Behav Biomed Mater. 2015;41:13–22.
Hupfer A, Brichkina A, Koeniger A, Keber C, Denkert C, Pfefferle P, et al. Matrix stiffness drives stromal autophagy and promotes formation of a protumorigenic niche. Proc Natl Acad Sci USA. 2021;118:e2105367118.
Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eaaw5473.
Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–39.
Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309:1074–8.
Liu QP, Luo Q, Deng B, Ju Y, Song GB. Stiffer matrix accelerates migration of hepatocellular carcinoma cells through enhanced aerobic glycolysis via the MAPK-YAP signaling. Cancers. 2020;12:490.
Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Investig. 2003;112:1776–84.
Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP, et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352.
Dong Y, Zheng Q, Wang Z, Lin X, You Y, Wu S, et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J Hematol Oncol. 2019;12:112.
Alonso-Nocelo M, Raimondo TM, Vining KH, López-López R, de la Fuente M, Mooney DJ. Matrix stiffness and tumor-associated macrophages modulate epithelial to mesenchymal transition of human adenocarcinoma cells. Biofabrication. 2018;10:035004.
Millet M, Bollmann E, Ringuette Goulet C, Bernard G, Chabaud S, Huot MÉ, et al. Cancer-associated fibroblasts in a 3D engineered tissue model induce tumor-like matrix stiffening and EMT transition. Cancers. 2022;14:3810.
Arnold JM, Gu F, Ambati CR, Rasaily U, Ramirez-Pena E, Joseph R, et al. UDP-glucose 6-dehydrogenase regulates hyaluronic acid production and promotes breast cancer progression. Oncogene 2020;39:3089–101.
Gao X, Qiao X, Xing X, Huang J, Qian J, Wang Y, et al. Matrix stiffness-upregulated microRNA-17-5p attenuates the intervention effects of metformin on HCC invasion and metastasis by targeting the PTEN/PI3K/Akt pathway. Front Oncol. 2020;10:1563.
Lin CY, Tsai PH, Kandaswami CC, Lee PP, Huang CJ, Hwang JJ, et al. Matrix metalloproteinase-9 cooperates with transcription factor Snail to induce epithelial-mesenchymal transition. Cancer Sci. 2011;102:815–27.
Lachowski D, Cortes E, Rice A, Pinato D, Rombouts K, Del Rio Hernandez A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci Rep. 2019;9:7299.
Li X, Pan J, Liu T, Yin W, Miao Q, Zhao Z, et al. Novel TCF21(high) pericyte subpopulation promotes colorectal cancer metastasis by remodelling perivascular matrix. Gut. 2023;72:710–21.
Enomoto Y, Matsushima S, Shibata K, Aoshima Y, Yagi H, Meguro S, et al. LTBP2 is secreted from lung myofibroblasts and is a potential biomarker for idiopathic pulmonary fibrosis. Clin Sci. 2018;132:1565–80.
Xie Y, Martin KA. TCF21: flipping the phenotypic switch in SMC. Circ Res. 2020;126:530–2.
Kuermanbayi S, Yang Y, Zhao Y, Li Y, Wang L, Yang J, et al. In situ monitoring of functional activity of extracellular matrix stiffness-dependent multidrug resistance protein 1 using scanning electrochemical microscopy. Chem Sci. 2022;13:10349–60.
Muñoz NM, Williams M, Dixon K, Dupuis C, McWatters A, Avritscher R, et al. Influence of injection technique, drug formulation and tumor microenvironment on intratumoral immunotherapy delivery and efficacy. J Immunother Cancer. 2021;9:e001800.
Yoon CW, Pan Y, Wang Y. The application of mechanobiotechnology for immuno-engineering and cancer immunotherapy. Front Cell Dev Biol. 2022;10:1064484.
Cai E, Marchuk K, Beemiller P, Beppler C, Rubashkin MG, Weaver VM, et al. Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science. 2017;356:eaal3118.
Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P. B cells use mechanical energy to discriminate antigen affinities. Science. 2013;340:1587–90.
Huse M. Mechanical forces in the immune system. Nat Rev Immunol. 2017;17:679–90.
Chaudhuri O, Cooper-White J, Janmey PA, Mooney DJ, Shenoy VB. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature. 2020;584:535–46.
Saraswathibhatla A, Indana D, Chaudhuri O. Cell-extracellular matrix mechanotransduction in 3D. Nat Rev Mol Cell Biol. 2023;24:495–516.
Meng KP, Majedi FS, Thauland TJ, Butte MJ. Mechanosensing through YAP controls T cell activation and metabolism. J Exp Med. 2020;217.
Hallmann R, Zhang X, Di Russo J, Li L, Song J, Hannocks MJ, et al. The regulation of immune cell trafficking by the extracellular matrix. Curr Opin Cell Biol. 2015;36:54–61.
Peranzoni E, Rivas-Caicedo A, Bougherara H, Salmon H, Donnadieu E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell Mol Life Sci. 2013;70:4431–48.
Freeman P, Mielgo A. Cancer-associated fibroblast mediated inhibition of CD8+ cytotoxic T cell accumulation in tumours: mechanisms and therapeutic opportunities. Cancers. 2020;12:2687.
Kato T, Noma K, Ohara T, Kashima H, Katsura Y, Sato H, et al. Cancer-associated fibroblasts affect intratumoral CD8(+) and FoxP3(+) T cells via IL6 in the tumor microenvironment. Clin Cancer Res. 2018;24:4820–33.
Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. 2018;9:948.
Jacobs J, Deschoolmeester V, Zwaenepoel K, Flieswasser T, Deben C, Van den Bossche J, et al. Unveiling a CD70-positive subset of cancer-associated fibroblasts marked by pro-migratory activity and thriving regulatory T cell accumulation. Oncoimmunology. 2018;7:e1440167.
Zhao X, Ding L, Lu Z, Huang X, Jing Y, Yang Y, et al. Diminished CD68(+) cancer-associated fibroblast subset induces regulatory T-cell (Treg) infiltration and predicts poor prognosis of oral squamous cell carcinoma patients. Am J Pathol. 2020;190:886–99.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422.
Pakravan K, Mossahebi-Mohammadi M, Ghazimoradi MH, Cho WC, Sadeghizadeh M, Babashah S. Monocytes educated by cancer-associated fibroblasts secrete exosomal miR-181a to activate AKT signaling in breast cancer cells. J Transl Med. 2022;20:559.
Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, et al. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019;10:273.
Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318:154–61.
Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C, et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35–48.
Rømer AMA, Thorseth ML, Madsen DH. Immune modulatory properties of collagen in cancer. Front Immunol. 2021;12:791453.
Bougherara H, Mansuet-Lupo A, Alifano M, Ngô C, Damotte D, Le frère-Belda MA, et al. Real-time imaging of resident T cells in human lung and ovarian carcinomas reveals how different tumor microenvironments control T lymphocyte migration. Front Immunol. 2015;6:500.
Nicolas-Boluda A, Vaquero J, Vimeux L, Guilbert T, Barrin S, Kantari-Mimoun C, et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. Elife. 2021;10.
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11:5120.
Huang JY, Cheng YJ, Lin YP, Lin HC, Su CC, Juliano R, et al. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: the role of tenascin-C in immune suppression. J Immunol. 2010;185:1450–9.
Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolytics. 2022;25:69–77.
Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018;15:366–81.
Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal protein betaig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut. 2019;68:693–707.
Carmona-Rodríguez L, Martínez-Rey D, Fernández-Aceñero MJ, González-Martín A, Paz-Cabezas M, Rodríguez-Rodríguez N, et al. SOD3 induces a HIF-2alpha-dependent program in endothelial cells that provides a selective signal for tumor infiltration by T cells. J Immunother Cancer. 2020;8:e000432.
O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189:1330–9.
Chin MHW, Norman MDA, Gentleman E, Coppens MO, Day RM. A hydrogel-integrated culture device to interrogate T cell activation with physicochemical cues. ACS Appl Mater Interfaces. 2020;12:47355–67.
Wang Y, Yang H, Jia A, Wang Y, Yang Q, Dong Y, et al. Dendritic cell Piezo1 directs the differentiation of T(H)1 and T(reg) cells in cancer. Elife. 2022;11:e79957.
Lim AR, Rathmell WK, Rathmell JC. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. Elife. 2020;9:e55185.
Kuczek DE, Larsen AMH, Thorseth ML, Carretta M, Kalvisa A, Siersbæk MS, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. 2019;7:68.
Zhao R, Zhou X, Khan ES, Alansary D, Friedmann KS, Yang W, et al. Targeting the microtubule-network rescues CTL killing efficiency in dense 3D matrices. Front Immunol. 2021;12:729820.
Castiglioni A, Yang Y, Williams K, Gogineni A, Lane RS, Wang AW, et al. Combined PD-L1/TGFbeta blockade allows expansion and differentiation of stem cell-like CD8 T cells in immune excluded tumors. Nat Commun. 2023;14:4703.
Chen M, Zhang Y, Zhou P, Liu X, Zhao H, Zhou X, et al. Substrate stiffness modulates bone marrow-derived macrophage polarization through NF-kappaB signaling pathway. Bioact Mater. 2020;5:880–90.
Chuang YC, Chang HM, Li CY, Cui Y, Lee CL, Chen CS. Reactive oxygen species and inflammatory responses of macrophages to substrates with physiological stiffness. ACS Appl Mater Interfaces. 2020;12:48432–41.
Bachy S, Wu Z, Gamradt P, Thierry K, Milani P, Chlasta J, et al. betaig-h3-structured collagen alters macrophage phenotype and function in pancreatic cancer. iScience. 2022;25:103758.
Wang S, Chen L, Liu W. Matrix stiffness-dependent STEAP3 coordinated with PD-L2 identify tumor responding to sorafenib treatment in hepatocellular carcinoma. Cancer Cell Int. 2022;22:318.
Erpenbeck L, Gruhn AL, Kudryasheva G, Günay G, Meyer D, Busse J, et al. Effect of adhesion and substrate elasticity on neutrophil extracellular trap formation. Front Immunol. 2019;10:2320.
Zhong H, Lu W, Tang Y, Wiel C, Wei Y, Cao J, et al. SOX9 drives KRAS-induced lung adenocarcinoma progression and suppresses anti-tumor immunity. Oncogene. 2023;42:2183–2194.
Mennens SFB, Bolomini-Vittori M, Weiden J, Joosten B, Cambi A, van den Dries K. Substrate stiffness influences phenotype and function of human antigen-presenting dendritic cells. Sci Rep. 2017;7:17511.
Jiang X, Hu J, Wu Z, Cafarello ST, Di Matteo M, Shen Y, et al. Protein phosphatase 2A mediates yap activation in endothelial cells upon VEGF stimulation and matrix stiffness. Front Cell Dev Biol. 2021;9:675562.
Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci USA. 2008;105:11305–10.
Kniazeva E, Putnam AJ. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am J Physiol Cell Physiol. 2009;297:C179–87.
Feng F, Feng X, Zhang D, Li Q, Yao L. Matrix stiffness induces pericyte-fibroblast transition through YAP activation. Front Pharm. 2021;12:698275.
Davidson MD, Song KH, Lee MH, Llewellyn J, Du Y, Baker BM, et al. Engineered fibrous networks to investigate the influence of fiber mechanics on myofibroblast differentiation. ACS Biomater Sci Eng. 2019;5:3899–908.
Almici E, Arshakyan M, Carrasco JL, Martínez A, Ramírez J, Enguita AB, et al. Quantitative image analysis of fibrillar collagens reveals novel diagnostic and prognostic biomarkers and histotype-dependent aberrant mechanobiology in lung cancer. Mod Pathol. 2023;36:100155.
Azadi S, Aboulkheyr Es H, Razavi Bazaz S, Thiery JP, Asadnia M, Ebrahimi Warkiani M. Upregulation of PD-L1 expression in breast cancer cells through the formation of 3D multicellular cancer aggregates under different chemical and mechanical conditions. Biochim Biophys Acta Mol Cell Res. 2019;1866:118526.
Chen Y, Zhao M, Zhang L, Shen D, Xu X, Yi Q, et al. SNF5, a core subunit of SWI/SNF complex, regulates melanoma cancer cell growth, metastasis, and immune escape in response to matrix stiffness. Transl Oncol. 2022;17:101335.
Miyazawa A, Ito S, Asano S, Tanaka I, Sato M, Kondo M, et al. Regulation of PD-L1 expression by matrix stiffness in lung cancer cells. Biochem Biophys Res Commun. 2018;495:2344–9.
Ding X, Wang L, Zhang X, Xu J, Li P, Liang H, et al. The relationship between expression of PD-L1 and HIF-1alpha in glioma cells under hypoxia. J Hematol Oncol. 2021;14:92.
Yuan G, Xie F, Song Y, Li Q, Li R, Hu X, et al. Hepatic tumor stiffness measured by shear wave elastography is prognostic for HCC progression following treatment with anti-PD-1 antibodies plus lenvatinib: a retrospective analysis of two independent cohorts. Front Immunol. 2022;13:868809.
Li YJ, Wu JY, Hu XB, Ding T, Tang T, Xiang DX. Biomimetic liposome with surface-bound elastase for enhanced tumor penetration and chemo-immumotherapy. Adv Health Mater. 2021;10:e2100794.
Zhang C, Xu M, Zeng Z, Wei X, He S, Huang J, et al. A polymeric extracellular matrix nanoremodeler for activatable cancer photo-immunotherapy. Angew Chem Int Ed Engl. 2023;135:e202217339.
Datta J, Dai X, Bianchi A, De Castro Silva I, Mehra S, Garrido VT, et al. Combined MEK and STAT3 inhibition uncovers stromal plasticity by enriching for cancer-associated fibroblasts with mesenchymal stem cell-like features to overcome immunotherapy resistance in pancreatic cancer. Gastroenterology. 2022;163:1593–612.
Jensen C, Madsen DH, Hansen M, Schmidt H, Svane IM, Karsdal MA, et al. Non-invasive biomarkers derived from the extracellular matrix associate with response to immune checkpoint blockade (anti-CTLA-4) in metastatic melanoma patients. J Immunother Cancer. 2018;6:152.
Hurkmans DP, Jensen C, Koolen SLW, Aerts J, Karsdal MA, Mathijssen RHJ, et al. Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J Immunother Cancer. 2020;8:e001193.
Ye Y, Kuang X, Xie Z, Liang L, Zhang Z, Zhang Y, et al. Small-molecule MMP2/MMP9 inhibitor SB-3CT modulates tumor immune surveillance by regulating PD-L1. Genome Med. 2020;12:83.
Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–81.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–8.
Manfredi F, Cianciotti BC, Potenza A, Tassi E, Noviello M, Biondi A, et al. TCR redirected T cells for cancer treatment: achievements, hurdles, and goals. Front Immunol. 2020;11:1689.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69.
Yeow YL, Kotamraju VR, Wang X, Chopra M, Azme N, Wu J, et al. Immune-mediated ECM depletion improves tumour perfusion and payload delivery. EMBO Mol Med. 2019;11:e10923.
Ishikawa T, Kokura S, Enoki T, Sakamoto N, Okayama T, Ideno M, et al. Phase I clinical trial of fibronectin CH296-stimulated T cell therapy in patients with advanced cancer. PLoS One. 2014;9:e83786.
Zhang Y, Pei P, Zhou H, Xie Y, Yang S, Shen W, et al. Nattokinase-mediated regulation of tumor physical microenvironment to enhance chemotherapy, radiotherapy, and CAR-T therapy of solid tumor. ACS Nano. 2023;17:7475–86.
Martín-Otal C, Lasarte-Cia A, Serrano D, Casares N, Conde E, Navarro F, et al. Targeting the extra domain A of fibronectin for cancer therapy with CAR-T cells. J Immunother Cancer. 2022;10:e004479.
Ahn S, Lee IH, Kang S, Kim D, Choi M, Saw PE, et al. Gold nanoparticles displaying tumor-associated self-antigens as a potential vaccine for cancer immunotherapy. Adv Health Mater. 2014;3:1194–9.
Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA. 2019;116:7624–31.
Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–66.
Lo A, Wang LCS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75:2800–10.
Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611–20.
Gulati P, Rühl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, et al. Aberrant Lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. 2018;24:3981–93.
Santos Apolonio J, Lima de Souza Gonçalves V, Cordeiro Santos ML, Silva Luz M, Silva Souza JV, Rocha Pinheiro SL, et al. Oncolytic virus therapy in cancer: a current review. World J Virol. 2021;10:229–55.
Lin D, Shen Y, Liang T. Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct Target Ther. 2023;8:156.
Yokoda R, Nagalo B, Vernon B, Oklu R, Albadawi H, DeLeon T, et al. Oncolytic virus delivery: from nano-pharmacodynamics to enhanced oncolytic effect. Oncolytic Virother. 2017;6:39–49.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–62.
Bazan-Peregrino M, Garcia-Carbonero R, Laquente B, Álvarez R, Mato-Berciano A, Gimenez-Alejandre M, et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J Immunother Cancer. 2021;9:e003254.
Kiyokawa J, Kawamura Y, Ghouse SM, Acar S, Barçın E, Martínez-Quintanilla J, et al. Modification of extracellular matrix enhances oncolytic adenovirus immunotherapy in glioblastoma. Clin Cancer Res. 2021;27:889–902.
Choi IK, Lee YS, Yoo JY, Yoon AR, Kim H, Kim DS, et al. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010;17:190–201.
Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98:1482–93.
Zhao Y, Baldin AV, Isayev O, Werner J, Zamyatnin AA Jr., Bazhin AV. Cancer vaccines: antigen selection strategy. Vaccines 2021;9:85.
Hu Y, Lin L, Chen J, Maruyama A, Tian H, Chen X. Synergistic tumor immunological strategy by combining tumor nanovaccine with gene-mediated extracellular matrix scavenger. Biomaterials. 2020;252:120114.
Yasunaga M, Manabe S, Tarin D, Matsumura Y. Cancer-stroma targeting therapy by cytotoxic immunoconjugate bound to the collagen 4 network in the tumor tissue. Bioconjug Chem. 2011;22:1776–83.
Huijbers EJM, Ringvall M, Femel J, Kalamajski S, Lukinius A, Åbrink M, et al. Vaccination against the extra domain-B of fibronectin as a novel tumor therapy. FASEB J. 2010;24:4535–44.
Van Limbergen EJ, Hoeben A, Lieverse RIY, Houben R, Overhof C, Postma A, et al. Toxicity of L19-interleukin 2 combined with stereotactic body radiation therapy: a phase 1 study. Int J Radiat Oncol Biol Phys. 2021;109:1421–30.
Poli GL, Bianchi C, Virotta G, Bettini A, Moretti R, Trachsel E, et al. Radretumab radioimmunotherapy in patients with brain metastasis: a 124I-L19SIP dosimetric PET study. Cancer Immunol Res. 2013;1:134–43.
Loeffler M. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Investig. 2006;116:1955–62.
Shin H, Kim Y, Jon S. Nanovaccine displaying immunodominant T cell epitopes of fibroblast activation protein is effective against desmoplastic tumors. ACS Nano. 2023;17:10337–10352.
Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9:1639–47.
Biragyn A, Schiavo R, Olkhanud P, Sumitomo K, King A, McCain M, et al. Tumor-associated embryonic antigen-expressing vaccines that target CCR6 elicit potent CD8+ T cell-mediated protective and therapeutic antitumor immunity. J Immunol. 2007;179:1381–8.
Kelly G, Milligan JJ, Mastria EM, Kim S, Zelenetz SR, Dobbins J, et al. Intratumoral delivery of brachytherapy and immunotherapy by a thermally triggered polypeptide depot. J Control Release. 2022;343:267–76.
Liu H, Lei D, Li J, Xin J, Zhang L, Fu L, et al. MMP-2 inhibitor-mediated tumor microenvironment regulation using a sequentially released bio-nanosystem for enhanced cancer photo-immunotherapy. ACS Appl Mater Interfaces. 2022;14:41834–50.
Yuan CS, Teng Z, Yang S, He Z, Meng LY, Chen XG, et al. Reshaping hypoxia and silencing CD73 via biomimetic gelatin nanotherapeutics to boost immunotherapy. J Control Release. 2022;351:255–71.
Yang Y, Ge S, Song Z, Zhao A, Zhao L, Hu Z, et al. A novel self-assembled epitope peptide nanoemulsion vaccine targeting nasal mucosal epithelial cell for reinvigorating CD8(+) T cell immune activity and inhibiting tumor progression. Int J Biol Macromol. 2021;183:1891–902.
Malfanti A, Bausart M, Vanvarenberg K, Ucakar B, Préat V. Hyaluronic acid-antigens conjugates trigger potent immune response in both prophylactic and therapeutic immunization in a melanoma model. Drug Deliv Transl Res. 2023;13:2550–2567.
Brauchle E, Kasper J, Daum R, Schierbaum N, Falch C, Kirschniak A, et al. Biomechanical and biomolecular characterization of extracellular matrix structures in human colon carcinomas. Matrix Biol. 2018;68-69:180–93.
Ebelt ND, Zamloot V, Zuniga E, Passi KB, Sobocinski LJ, Young CA, et al. Collagenase-expressing salmonella targets major collagens in pancreatic cancer leading to reductions in immunosuppressive subsets and tumor growth. Cancers. 2021;13:3565.
Redman JM, Friedman J, Robbins Y, Sievers C, Yang X, Lassoued W, et al. Enhanced neoepitope-specific immunity following neoadjuvant PD-L1 and TGF-beta blockade in HPV-unrelated head and neck cancer. J Clin Investig. 2022;132:e172059.
Cho BC, Daste A, Ravaud A, Salas S, Isambert N, McClay E, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in advanced squamous cell carcinoma of the head and neck: results from a phase I cohort. J Immunother Cancer. 2020;8:e000664.
Lin CC, Doi T, Muro K, Hou MM, Esaki T, Hara H, et al. Bintrafusp Alfa, a bifunctional fusion protein targeting TGF beta and PD-L1, in patients with esophageal squamous cell carcinoma: results from a phase 1 cohort in Asia. Target Oncol. 2021;16:447–59.
Tan B, Khattak A, Felip E, Kelly K, Rich P, Wang D, et al. Bintrafusp Alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with esophageal adenocarcinoma: results from a phase 1 cohort. Target Oncol. 2021;16:435–46.
Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH, et al. Bintrafusp Alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in second-line treatment of patients With NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol. 2020;15:1210–22.
Kang YK, Bang YJ, Kondo S, Chung HC, Muro K, Dussault I, et al. Safety and tolerability of bintrafusp Alfa, a bifunctional fusion protein targeting TGFbeta and PD-L1, in Asian patients with pretreated recurrent or refractory gastric cancer. Clin Cancer Res. 2020;26:3202–10.
Yoo C, Oh DY, Choi HJ, Kudo M, Ueno M, Kondo S, et al. Phase I study of bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with pretreated biliary tract cancer. J Immunother Cancer. 2020;8:e000564.
Strauss J, Gatti-Mays ME, Cho BC, Hill A, Salas S, McClay E, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-beta and PD-L1, in patients with human papillomavirus-associated malignancies. J Immunother Cancer. 2020;8:e001395.
Spira A, Wertheim MS, Kim EJ, Tan B, Lenz HJ, Nikolinakos P, et al. Bintrafusp Alfa: a bifunctional fusion protein targeting PD-L1 and TGF-beta, in patients with pretreated colorectal cancer: results from a phase I trial. Oncologist. 2023;28:e124–e127.
Tolcher AW, Gordon M, Mahoney KM, Seto A, Zavodovskaya M, Hsueh CH, et al. Phase 1 first-in-human study of dalutrafusp alfa, an anti-CD73-TGF-beta-trap bifunctional antibody, in patients with advanced solid tumors. J Immunother Cancer. 2023;11:e005267.
Rocconi RP, Grosen EA, Ghamande SA, Chan JK, Barve MA, Oh J, et al. Gemogenovatucel-T (Vigil) immunotherapy as maintenance in frontline stage III/IV ovarian cancer (VITAL): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Oncol. 2020;21:1661–72.
Giaccone G, Bazhenova LA, Nemunaitis J, Tan M, Juhász E, Ramlau R, et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur J Cancer. 2015;51:2321–9.
Liu D, Zhou J, Wang Y, Li M, Jiang H, Liu Y, et al. Bifunctional anti-PD-L1/TGF-betaRII agent SHR-1701 in advanced solid tumors: a dose-escalation, dose-expansion, and clinical-expansion phase 1 trial. BMC Med. 2022;20:408.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–66.
Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. 2022;28:724–34.
Wang-Gillam A, Lim KH, McWilliams R, Suresh R, Lockhart AC, Brown A, et al. Defactinib, pembrolizumab, and gemcitabine in patients with advanced treatment refractory pancreatic cancer: a phase I dose escalation and expansion study. Clin Cancer Res. 2022;28:5254–62.
Melero I, Tanos T, Bustamante M, Sanmamed MF, Calvo E, Moreno I, et al. A first-in-human study of the fibroblast activation protein-targeted, 4-1BB agonist RO7122290 in patients with advanced solid tumors. Sci Transl Med. 2023;15:eabp9229.
Zhou M, Zhu S, Xu C, Liu B, Shen J. A phase Ib/II study of BLU-554, a fibroblast growth factor receptor 4 inhibitor in combination with CS1001, an anti-PD-L1, in patients with locally advanced or metastatic hepatocellular carcinoma. Investig N Drugs. 2023;41:162–7.
Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338–48.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81901006, 82372905), Young Top-notch Talent of Pearl River Talent Plan (0920220228), Guangdong Provincial Science and Technology Project Foundation (2022A0505050038), and Science Research Cultivation Program of Stomatological Hospital, Southern Medical University (PY2020002, PY2022019).
Author information
Authors and Affiliations
Contributions
LC, XZ, and ZM conceived of the presented idea. LC, ZM, XZ, YL, and PL wrote the manuscript. LC and XZ provided the guidance throughout the revision of this manuscript. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: Francesca Bernassola.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mai, Z., Lin, Y., Lin, P. et al. Modulating extracellular matrix stiffness: a strategic approach to boost cancer immunotherapy. Cell Death Dis 15, 307 (2024). https://doi.org/10.1038/s41419-024-06697-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06697-4
