
Abstract
Recently, heterozygous loss-of-function NFKB1 variants were identified as the primary cause of common variable immunodeficiency (CVID) in the European population. However, pathogenic NFKB1 variants have never been reported in the Japanese population. We present a 29-year-old Japanese woman with CVID. A novel variant, c.136 C > T, p.(Gln46*), was identified in NFKB1. Her mother and daughter carried the same variant, demonstrating the first Japanese pedigree with an NFKB1 pathogenic variant.
Similar content being viewed by others

The most common primary immunodeficiency disease characterized by defective antibody production is common variable immunodeficiency (CVID)1. CVID is a clinically and genetically heterogeneous disorder characterized by impaired antibody production and recurrent infections. However, the genetic cause of CVID in the majority of patients remains unknown. Recent large-scale whole-genome sequencing analysis identified NFKB1 as the most common causative gene of CVID, accounting for 4% of CVID cases in a predominantly European population2. No pathogenic variants in NFKB1 have been reported in the Japanese population to date.
The proband was a 29-year-old female patient whose medical history consisted only of an annual fever and rash. She exhibited mild flexion restriction in the right middle finger and left ring finger at the proximal interphalangeal joint since her late teens, with occasional pain in the wrist and ankle. Her 54-year-old mother had rheumatoid arthritis since the age of 52 and was prescribed methotrexate. Her 57-year-old father, two brothers aged 32 and 25 years, and three-year-old daughter were healthy (Fig. 1A). When she visited a dermatologist for two weeks of fever and stomatitis, she was found to have marked hypogammaglobulinemia, with immunoglobulin G (IgG), IgG2, IgA, and IgM levels less than 0.06, 0, 0, and less than 0.02 g/L, respectively. The absolute lymphocyte count was 1740 cells/μl, while the white blood cell count was 7900 cells/μl. The percentage of CD19+ B cells within the total lymphocyte population was markedly reduced to 0.2%. According to flow cytometric analyses, the counts of other peripheral blood lymphocyte subsets were within the normal ranges as follows: CD3+ T cells, 92%; CD4+ T cells, 50%; CD8+ T cells, 36%; and CD16+ CD56+ natural killer (NK) cells, 7.6%.

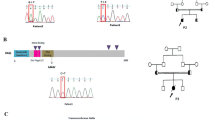
A Pedigree of the family. The proband’s 54-year-old mother (I:2) suffered from rheumatoid arthritis. Her 57-year-old father (I:1), two brothers aged 32 (II:1) and 25 years (II:4), and a 3-year-old daughter (III:1) were healthy. B Flow cytometric analysis of B-cell subsets in the peripheral blood of the proband’s daughter. (B-a) Flow cytometric analysis of CD19+ CD20+ B cells among total lymphocytes. (B-b) Flow cytometric analysis of IgD+ CD27− naive B cells, IgD+ CD27+ IgM memory B cells, and IgD− CD27+ switched B cells within the subset of CD19+ CD20+ B cells. C Electropherogram of Sanger sequencing revealing the same variant, c.136 C > T (p.Gln46*), in NFKB1 in the proband, mother, and daughter. D Schematic representation of the protein domains and previously reported genetic variants in NFKB1. The black and white arrowheads indicate this patient and previously reported patients, respectively. RHD Rel homology domain, GRR glycine-rich region, DD death domain.
The results of 3H-thymidine incorporation assays induced by phytohemagglutinin and concanavalin A were normal at 76,100 cpm (stimulation index: 127–456) and 69,700 cpm (stimulation index: 127–456), respectively. A Cr51 release assay was used to assess NK cytotoxicity, and the result of 22% was normal. Antibodies specific for Japanese encephalitis, measles, and rubella were all negative although the patient had been vaccinated against these antigens. Abdominal ultrasonography revealed no hepatosplenomegaly or intra-abdominal lymph node enlargement. Chest computed tomography revealed no thymoma suggestive of Good’s syndrome, and chronic airway inflammation due to repeated infections was not observed.
The patient was diagnosed with CVID based on a markedly reduced serum IgG level accompanied by decreased IgA and IgM, an absent antibody response to vaccinations, and the lack of other causes of immunodeficiency. The proband and her parents provided written informed consent for genetic testing following genetic counseling. The Medical Ethics Committee of Kobe University, Kobe, Japan (IRB number: B210180) approved this genetic study. Peripheral blood leukocytes were isolated from the proband and her parents. The QuickGene-Auto 12 S system (Wako Pure Chemical Industries, Ltd., Tokyo, Japan) was used to extract genomic DNA following the manufacturer’s instructions. An Illumina NextSeq 2000 (Illumina, San Diego, CA) and Twist Comprehensive Exome Panel (TWIST Bioscience, South San Francisco, CA) were used for whole-exome sequencing (WES).
A novel heterozygous nonsense variant of NFKB1 [NM_003998.4: c.136 C > T, p.(Gln46*)] was identified in the region encoding the N-terminal Rel homology domain (RHD) by WES; Sanger sequencing was subsequently performed in the proband and her mother (Fig. 1C). This variant was not reported in the Human Gene Mutation Database (https://www.hgvd.genome.med.kyoto-u.ac.jp/). The variant was classified as pathogenic (pathogenic very strong (PVS) 1 + pathogenic moderate (PM) 2 + pathogenic supporting (PP) 1) according to the American College of Medical Genetics and Genomics guidelines because the loss of NFKB1 function is known to cause CVID, and the variant identified in the proband and her mother was null.
Although the proband’s daughter was asymptomatic, she underwent an immunological examination because of the autosomal dominant inheritance pattern of NFKB1. The daughter had reduced IgG, IgG2, IgM, and IgA levels of 3.0 g/L, 0.41 g/L, 0.22 g/L, and 0.04 g/L, respectively. These values were more than 2 SDs below the age-specific normal ranges3. The absolute lymphocyte count was 5100 cells/μl, while the white blood cell count was 8300 cells/μl. The percentage of CD19+ B cells within the total lymphocyte population was 8.2%, and the absolute count was 420 cells/μl. The median percentage and absolute count of CD19+ B cells in 3-year-old children are 17% (10th–90th percentile: 9–31%) and 590 cells/μl (10th–90th percentile: 310–1130 cells/μl)4, respectively. Therefore, the daughter’s B-cell counts were not significantly lower than the age-specific normal values. The percentages (median and 10th–90th percentile of 3-year-old children4) of IgD+ CD27− naive cells, IgD+ CD27+ memory cells, and IgD− CD27+ switched memory cells among total B cells were 89.0% (74% and 63–86%), 7.6% (22% and 14–43%), and 1.8% (17% and 7–34%), respectively (Fig. 1B). These results indicated that impaired development from naive B cells to switched memory B cells was the cause of defective antibody production. Similarly, a reduced number of memory B cells in patients with NFKB1 haploinsufficiency has been reported2,5,6,7. Intriguingly, the IgG levels and percentage of CD19+ B cells within the total lymphocyte population in the proband’s mother were 8.2 g/dl and 11%, respectively, which were within the normal range despite the fact that she carried the same NFKB1 pathogenic variant. The daughter had hypogammaglobulinemia; thus, genetic testing was performed after obtaining written informed consent from her parents. The results revealed the same NFKB1 variant in the daughter (Fig. 1C).
Immunoglobulin replacement therapy for severe hypogammaglobulinemia and recurrent infections was initiated in the proband. Her fever resolved after initiating immunoglobulin replacement therapy.
Observation was chosen for the proband’s mother, who did not have overt hypogammaglobulinemia, and the proband’s daughter, who had hypogammaglobulinemia but was asymptomatic.
Here, we report a Japanese family with CVID harboring a novel c.136 C > T variant in NFKB1. To our knowledge, this is the first reported case of a pathogenic NFKB1 variant in a Japanese patient with CVID.
Nuclear factor kappa B (NFκB) is a transcription factor consisting of RelA (p65), RelB, C-Rel, NFκB1 (precursor p105/activator p50), and NFκB2 (precursor p100/activator p52). NFκB regulates more than 500 target genes and is involved in various signal transduction pathways, such as cell differentiation, survival, inflammatory response, and immunosuppression8. Loss of NFκB function is associated with CVID development, which is characterized by susceptibility to infection due to impaired development of antibody-producing mature B cells. The typical age for CVID diagnosis is 20–40 years2. Serum IgG levels and absolute B-cell numbers are variable2. Hypogammaglobulinemia is the most common manifestation of NFKB1 haploinsufficiency, followed by respiratory tract infections and abscess formation6. Autoimmune diseases have been reported6, which is consistent with the mother’s rheumatoid arthritis. Approximately 15% of patients develop malignancies6; thus, careful follow-up is needed. Other less common symptoms include gut involvement, hepatosplenomegaly, lymphadenopathy, recurrent or chronic diarrhea, and Epstein–Barr virus infection6.
Recent large-scale whole-genome sequencing analysis identified pathogenic NFKB1 variants in 16 of 390 (4%) patients with CVID in a European cohort2. Some genetic abnormalities involving NFKB29,10,11 have been identified in Japanese CVID patients12, although NFKB1 pathogenic variants have never been reported.
Figure 1D and Table 1 summarize previously reported NFKB1 pathogenic variants. NFKB1 consists of 24 exons (GenBank annotation NC_000004.11: 103422486-103538459) and encodes the 969 amino acid p105 and its shorter isoform 2 (968 amino acids). In order from the N-terminus, its structure consists of a Rel homology domain (RHD), a glycine-rich region, an ankyrin repeat, and a death domain (DD). The RHD mediates dimerization, specific protein inhibitor interactions, and DNA binding13. Most of the previously reported pathogenic variants were localized in the RHD (Fig. 1D). The variant identified in this patient was located on the N-terminal side among the previously reported variants, except for two large deletions (Fig. 1D).
NFKB1 exhibits an autosomal dominant inheritance pattern, but differences in symptoms among relatives with the same NFKB1 variant have been reported6,13,14.
The characteristics of the cohort of NFKB1 variant carriers indicate incomplete clinical penetrance even with advancing age. This is likely due to the involvement of other genetic, epigenetic, or environmental factors in CVID development. Individualized follow-up for each patient is required because of the variation in immunological and other manifestations even between individuals within the same pedigree harboring the same NFKB1 pathogenic variant. Additionally, our immunological study of the proband’s daughter provides valuable information regarding the early steps in the development of overt clinical disease.
We reported the first Japanese patient with the pathogenic NFKB1 variant [c.136 C > T, p.(Gln46*)]. Clinical manifestations are highly variable even among family members having the same NFKB1 variant. Genetic testing can be useful for managing patients with CVID.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3379.
References
Gathmann, B. et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 134, 116–126 (2014).
Tuijnenburg, P. et al. Loss-of-function nuclear factor kB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 142, 1285–1296 (2018).
Yamada, Y. Laboratory test values in children significantly depend on age: focus on alkaline phosphatase (ALP), alpha-fetoprotein (AFP), and immunoglobulins. Rinsho Byori 62, 795–801 (2014).
Garcia-Prat, M. et al. Extended immunophenotyping reference values in a healthy pediatric population. Cytom. B Clin. Cytom. 96, 223–233 (2019).
Schröder, C. et al. Late-onset antibody deficiency due to monoallelic alterations in NFKB1. Front. Imuunol. 10, 2618 (2019).
Dieli-Crimi, R. et al. Th1-skewed profile and excessive production of proinflammatory cytokines in a NFKB1-deficient patient with CVID and severe gastrointestinal manifestations. Clin. Immunol. 195, 49–58 (2018).
Kaustio, M. et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J. Allergy Clin. Immunol. 140, 782–796 (2017).
Lougaris, V. et al. Early and late B-cell developmental impairment in nuclear factor kappaB, subunit 1–mutated common variable immunodeficiency disease. J. Allergy Clin. Immunol. 139, 349–352 (2017).
Okamura, K. et al. Neutrophilic dermatosis associated with an NFKB2 mutation. Clin. Exp. Dermatol. 44, 350–352 (2019).
Nagai, M., Imai, Y. & Yamanishi, K. Psoriasiform dermatitis associated with common variable immunodeficiency 10 due to an Arg853* mutation in the NFKB2 gene. J. Dermatol. 46, e24–e26 (2019).
Wada, S. et al. Generalized pustular psoriasis that developed in a patient with an NFKB2 variant. J. Dtsch Dermatol Ges. 22, 118–120 (2024).
Okano, T. et al. Whole-exome sequencing-based approach for germline mutations in patients with inborn errors of immunity. J. Clin. Immunol. 40, 729–740 (2020).
Fliegauf, M. et al. Haploinsufficiency of the NF-kB1 subunit p50 in common variable immunodeficiency. Am. J. Hum. Genet. 97, 389–403 (2015).
Tuovinen, E. A. et al. Long-term follow up of families with pathogenic NFKB1 variants reveals incomplete penetrance and frequent inflammatory sequelae. Clin. Immunol. 246, 109181 (2023).
Keller, B. et al. Disturbed canonical nuclear factor of kappa light chain signaling in B cells of patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 139, 220–231 (2017).
Maffucci, P. et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front. Immunol. 7, 220 (2016).
Boztug, H. et al. NF-κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J. Clin. Immunol. 36, 533–540 (2016).
Gonzalez-Granado, L. I. et al. Acquired and innate immunity impairment and severe disseminated mycobacterium genavense infection in a patient with a NF-κB1 deficiency. Front. Immunol. 29, 3148 (2019).
Rae, W. et al. Autoimmunity/inflammation in a monogenic primary immunodeficiency cohort. Clin. Trans. Immunol. 6, e155 (2017).
Lougaris, V. et al. NFKB1 regulates human NK cell maturation and effector functions. Clin. Immunol. 175, 99–108 (2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nakatani, N., Tamura, A., Hanafusa, H. et al. A novel NFKB1 variant in a Japanese pedigree with common variable immunodeficiency. Hum Genome Var 11, 15 (2024). https://doi.org/10.1038/s41439-024-00271-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00271-2
