
Abstract
State-of-the-art lithium-ion batteries inevitably suffer from electrode corrosion over long-term operation, such as corrosion of Al current collectors. However, the understanding of Al corrosion and its impacts on the battery performances have not been evaluated in detail. The passivation, its breakdown, and corrosion of the Al resulted in the deterioration of the solid/solid interface and electrode integrity. Additionally, localized diffusion of F−/Al3+ brought the irreversible current detrimental to the Coulomb efficiency (1.14% loss). Eventually, the behavior led to extensive capacity damage (>20%) to battery performance until lifespan. During the battery cycling, the passivation layer greater than 20 nm was generated near the median voltage. When the charging voltage rose, the passivation layer was squeezed and deformed by the newly generated Al-F-O particles, resulting in stress corrosion cracks. The passivation layer peeled off, and the nano-passivation layer material was re-generated as the voltage continued to rise. The above results were repeated, and the Al matrix was continuously consumed. The passivity breakdown with localized corrosion was derived from ethylene carbonate adsorption, which was highly correlated to the charge voltages, especially at 4.4 V and 4.8 V. The results will serve as a benchmark for electrode corrosion of other advanced energy storage materials, which is crucial for electrode engineering and performance modulation using interfacial design.
Similar content being viewed by others


Introduction
One of the application dilemmas of lithium-ion batteries (LIBs) in portable electronic devices is the requirement to manage the physical-chemical stability of multiple external/internal components. The internal components all contain separators1,2, current collectors3,4,5,6,7,8, electrolytes9,10,11,12,13, and electrodes14,15,16,17,18. Due to the high energy density of LIBs, their application has also penetrated the automotive market in the form of plug-in hybrid and electric vehicles. However, for batteries to be an economical and reliable form of automotive energy storage, the next generation of higher energy-density LIBs must have high-cutoff voltages and structural stabilities. Herein, the internal component stability should be noticed and strengthened less prone to degradation or failures19 (e.g., corrosion the of current collector8,20,21,22,23,24,25,26 and spoilage of electrode materials24,16,27).
Al/Cu are the most concerned materials for the cathode/anode current collectors in lithium-based and other rechargeable energy batteries attributed to their cost-efficiencies, high electrical conductivity, and chemical/electrochemical inertness28,29. However, metal corrosion degrades metallic materials and their structures, which behave as Cu oxidation and Al dissolution for LIBs’ current collectors. Degradation of Cu is an important aspect, as this component contributes significantly to the battery weight and cost. The inertness of Al originates from the original oxide film, which is composed of two layers, i.e., hard crystalline hydro-soft aluminite (Al2O3·H2O) with fine holes and the middle layer is a dense amorphous alumina (Al2O3) barrier layer30. Inevitably, the Al substrate will be exposed to aggressive electrolytes and be attacked despite its natural oxide film4,22,31,32,33,34,35. The attacking effects from liquid organic electrolytes (i.e., ester-based electrolytes) are distinct from those in aqueous environments36. The former mechanism exposure is indispensable for the development of energy storage materials. The subsequent corrosion may be related to the contaminates and side reactions/products of the electrolytes, such as HF generated from LiPF6 hydrolysis in the presence of a trace amount of water, LiTFSI, and LiFSI20,22,37,38,39. The parasitic corrosive reactions of the electrolyte are reflected as undesired interfacial interactions, which contain the chemical corrosion of current collectors and the dissolution of electrodes. The latter dissolution has been examined in several literature16,40,41,42. In contrast to the latter, the chemical/electrochemical corrosion and failure of current collectors at multiple charge voltages need to be evaluated corresponding to the battery performance, especially at >4.5 V. Thus, it is urgent to determine the corrosion influence on battery performance and the mechanisms of Al corrosion at the commercial LIBs’ level.
Limited efforts have been devoted to understanding the corrosion behaviors of Al in the commercially available LIBs’ electrolytes, and the coupling impacts of anions and solvents of the electrolytes on Al corrosion have not been showcased. Taeho Yoon et al.4 came up with the passivation of Al mainly owing to an electrochemical reaction rather than HF from LiPF6 hydrolysis (the electrolyte: 1.3 M LiPF6 in ethylene carbonate (EC)/ethyl methyl carbonate (EMC)/diethyl carbonate (DEC), v/v/v = 3:2:5). These researchers et al.43,44 persisted that the corrosion of the Al was initiated by the oxidation of uncoordinated solvent molecules, which generated the organic radical cations. Zhu et al.45 used the Al current collectors from spent LIBs and indicated the high rate performances of LiNi0.6Mn0.2Co0.2O2 on reused Al deteriorating a lot in 1 M LiPF6 + EC: DMC (dimethyl carbonate) (3:7 v/v) + VC 1 wt%. Al current collectors of commercial LIBs continually will experience the impact of interfacial parasitic reactions during battery service. It is necessary and rational to evaluate the ratio of corrosion to performance attenuation by using these current collectors and the impacts of electrode potentials on passivation/corrosion. Otherwise, the cognition of Al passivation/corrosion in ester-based electrolytes will shed light on the stability application of Al current collectors in sodium/potassium-ion batteries.
Here, we compared the stability performance between reclaimed Al current collector (RA) after the whole service of LiCoO2 batteries and fresh Al (FA) foils, which could elucidate the multiple changes of Al after battery operation. Moreover, using LiCoO2 as the cathode material to assemble the LiCoO2||Li half-cells, and the electrochemical performances of the electrodes using FA and RA as current collectors were evaluated. The mentioned detection could shed light on the deterioration extent of cathode material performances from Al changing. Subsequently, the influences of the potentials applied on Al (cathodes) were detected to verify the passivation/corrosion processes, which is valuable to the development of high-voltage LIBs. The adsorption effects of electrolyte components on Al substrate were simulated using density functional theory (DFT), which helped elucidate the competitive adsorption and underlying oxidation processes of multiple molecules. We aim to reveal Al corrosion and resulting battery performance degradation in LIBs, which is significant toward the understanding of the high voltage stability of Al current collectors in various energy storage materials.
Results
Characterization of reclaimed Al and electrochemical performance evaluation
The handling process of reclaimed Al was depicted in Fig. 1a. The morphology of RA was inhomogeneous up to the battery lifespan (Fig. 1b, c and Supplementary Figs. 1 and 2), and the roughness of Al was randomly distributed. Stratiform layers, cracks, and pits were generated and accompanied by the presence of Al/O/F complexes on the surface of RA (Fig. 1e and Supplementary Figs. 1 and 2). Especially, the RA surfaces in Supplementary Fig. 1 became porous and reticulate, and there were surface faults on the RA surface which can be evidenced by layer stages of several profiles (Supplementary Fig. 2). The XRD patterns of RA were the (311), (220), and (111) orientations (Fig. 1d, consulting Al PDF#04-0787). Obvious pits were observed in the RA (Supplementary Fig. 1d, e). Studies have shown that such perforations help reduce the weight of the current collector and facilitate the transfer of ions between the two sides of the current collector46,47. This would help to balance the circulation and power density. But for the battery which has already been in the process of charge and discharge, the perforation of the current collector was clear to the deterioration of electron transfer. This uneven punching pattern would directly reduce the electrical conductivity3, which means excessive surface passivation/localized corrosion of the current collector.

a Schematics of RA current collectors from lithium-ion batteries. b, c AFM and (d) XRD characterization of RA. XPS high-resolution profiles of Li 1s, Al 2p, O 1s, and F 1s on the (e) RA and (f) FA surfaces.
The wide XPS spectrums of Al foils demonstrated that the binding energies (BE) of Li 1s, Al 2p, P 2p, O 1s, and F1s are 56.2, 76.8, 134.5, 533.4, and 687.2 eV, respectively. As shown in Fig. 1e, the BEs of Al 2p, O 1s, and F 1s were slightly shifted compared with the pure elements48,49. This may arise due to the interaction between the F element of AlF3 and O, forming a chemical bond that leads to a change in the outer electronic structure.
The initial surface layers of the FA were Al-F/Al-O-F/Li-F in Fig. 1f, which was distinct from that of RA (i.e., Al-F/Al-O-F) suffering from battery operation. In contrast to RA, there was a trace of LiF on the surface of FA which was passivated (Fig. 1f). This might be attributed to electrolyte decomposition and LiF generation in internal sites of the passivation layer at the initial cycling. In the waste batteries, the Al-F/Al-O-F layers were spontaneously generated after the continuous dissolution of Al until the depth of 20 nm, which meant the parasitic reactions occurred at the interfaces during the battery charge/discharge. These structures were loose and porous (Supplementary Fig. 1)30, and the AlF3 layer with obvious passivation protection in the contrast peak area was significantly reduced. In the other two states, the BEs of Al surface passivation were slightly offset (Fig. 1f Al 2p). The displacement of the O1s peak in the direction of sputtering depth was due to the change of oxide proportion. In addition, a large shift in the peak of F 1s toward higher eV was observed, which contributed to the correlation with more electronegative ions’ injection.
In addition to the LiF on the FA surface, the oxidation/passivation layer still existed at 20 nm. However, there was no Al BE peak in the Al 2p of RA, which speculated that it arose from the thick and inhomogeneous passivation layer (thickness >20 nm) on the Al structure (Fig. 1e). This phenomenon was related to the F−/O2− inward penetration and inner interfacial Al outward dissolution. In short, the predominant passivation layer was different for FA and RA, which were fluorides (binding energy F 1s (AlF3) at about 687.8 eV, and Al-O-F at 686.6 eV) and oxides (binding energy of Al2O3 at about 74.5 eV)4,48,49, respectively. The phenomena indicated that the Al current collector may experience fluoride passivation, passivity breakdown, and localized corrosion and oxidation during battery operation.
Accordingly, the electrochemical performances of RA and FA were detected in detail (Fig. 2 and Supplementary Fig. 3). The oxidation current was ascribed to the corrosion of Al and the electrolyte decomposition. For the 1st cyclic voltammogram (CV) of Al||Li cells, the anodic current of FA started with 3.76 V versus Li/Li+ of FA (Supplementary Fig. 3b). In contrast, the anodic dissolution of RA began with 3.89 V versus Li/Li+ and was accompanied by a higher anodic current (Supplementary Fig. 3a). The phenomenon corresponded to the distinction of the native passivation layer between RA and FA. Reactions of Al electrodes were shown in Eqs. (1) and (2) as follows:

a Amperometric i-t curves of FA||Li and RA||Li for 4 V-24 h. The current collectors were cycled 3 times with a scanning rate of 10 mV s−1. EIS of FA/RA current collectors in 1 M LiPF6- FEC + EC + DEC (v/v/v, 1: 3: 6) at (b) OCP before 3rd CV, (c) OCP after 3rd CV, (d) 4.6 V before 3rd CV, (e) 4.6 V after 3rd CV. f Equivalent circuit.
Ulteriorly, the anodic dissolution of FA was terminated within proceeding cycling, which was probably ascribed to the accompanied passivation (Fig. 1f). The anodic current density of RA||Li increased dramatically beginning with the 5th cycle, which signified the localized corrosion of the interfacial layers. Amperometric i-t curves of FA||Li and RA||Li for 4 V-24 h also elucidated that the RA would be corroded continually (Fig. 2a). At the same time, the current (electric quantity) of RA at 4 V meant the irreversible capacity for the batteries. The phenomenon verified that the passivation layers on RA were inhomogeneous and the Al substrate was not hidden and protected by them. The localized corrosion may happen at the edge of stratiform layers and crevices.
The Nyquist plots of FA and RA (Fig. 2b–e) demonstrated less corrosion resistance of RA than that of FA after cycling. The electrochemical impedance spectrums (EIS) spectra were fitted, and as a result, the equivalent circuit could be obtained shown in Supplementary Table 2 and Fig. 2f. At high frequencies, the impedances of the interfaces presented resistance-constant phase element (CPE) characteristics50,51. The CPE was believed to be generated from the inhomogeneity of the electrode surface. Bode plots showed two characteristic frequencies, which represent two state variables. It proved that with the operation of the cell, the RA impedance would continually decrease, resulting the lower corrosion resistance (both charge transfer resistance and diffusion resistance). In addition, the decrease of Rct2 before and after CV meant that the passivation layer exposed to the electrolytes was corroded especially for the samples undergone CV. At the initial operation process, the values of R3 (diffusion resistance at the barrier layer) of 4.6 V state still presented that FA possesses higher Al3+ diffusion impedance than that of RA, while the increase of R3 at 4.6 V state (Fig. 2e) after CV indicated that the reconstructed passivation layer protects Al substrate. The above results indicated that RA still behaved high corrosion tendency attributing to deeply localized corrosion despite its passivation in the former.
Noting that the morphology of RA seemed to be corroded, the larger surface area corresponded to the lower charge transfer resistance (Rct) compared to that of FA. Also, the phenomenon would introduce more intricate parasitic reactions at the interface of solid phase and liquid electrolyte, especially for Al active electrodes. Interestingly, the 3rd cycling pushed the passivation of the Al surface at 4.6 V versus Li/Li+ (Fig. 2d, e). This was coincident with the results that fluoride participated in the passivation process. The fluorides were generated from the LiPF6 decomposition. The more exaggerated heterogeneity of the passivation layer, the more deteriorating corrosion-protecting effect corresponded to the substrate (Fig. 2a). The subsequent parasitic reactions in the inhomogeneous layers would result in excessive consumption and contamination of active substrate and electrolyte. Because Al foil would dissolve/consume and deteriorate the adherence between the electrode material and the current collector, which results in accelerating the decay of battery performance.
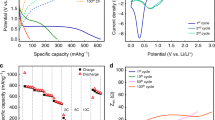
In LiCoO2||Li half cells with FA or RA as current collectors (Fig. 3a), the rate performances of RA degraded with capacity loss >20% compared with FA. In contrast to the current density of 5 C, the CRs (100th) of RA at 0.5 C were much lower than that of FA within 100 cycles (Fig. 3b). The phenomenon meant that the corrosion at the low current state was much more serious than that of the high one in 1 M LiPF6 + EC/DEC/FEC when the discharge level was consistent. Subversively, the results were different with NCM622 on reclaimed Al for 100th cycles, whose electrolyte selected 1 M LiPF6 in EC: DMC 3: 7 v/v + VC 1 wt.%45. However, the rate performances had the same trend. According to the EIS of LiCoO2||Li half cells, the LiCoO2 on RA presented higher interfacial resistance than that of FA (Fig. 3c, d, inner pictures of the Bode plots (log|z|−logf), and Supplementary Table 3). This phenomenon responded to the sluggish kinetics during progressive cycling. According to Fig. 3e, the Coulombic Efficiency of RA was in the range of 98.70% which was lower than 99.84% of FA. Combining Fig. 2a, it might stem from RA’s continuing anodic current of passivity breakdown and localized corrosion. The results meant that the corrosion of Al current collectors may deteriorate the specific capacity of LiCoO2||Li by more than 20% within the lifespan. The cycle number (200th~800th) indicated that LiCoO2||Li on FA suffered from capacity fading more critically than LiCoO2||Li on excessively passivated RA, which might originate from the distinction of Al structures corresponding to plane and corroded-porous three-dimensional structure.

a Rate performances of LiCoO2 on different Al foils. b The capacity retention of LiCoO2 on different Al foils at 0.5 C, 1 C, and 5 C after the 100th cycle. c, d EIS curves of different Al foils after the 3rd and 100th cycles, electrolyte: 1 M LiPF6 + EC/DEC/FEC. e Cycle performances of FA and RA at 1 C.
It is highly needed to elucidate the corrosive mechanism of the Al current collector in detail. Three factors should be considered: (1) the decomposition/oxidation of polar solvent molecules and salt anions; (2) the anodic dissolution of Al (Eq. (2)); and (3) the coupled reactions between (1) products and Al3+. The surface roughness of RA after cycling-recovery is different from that of FA, and the comparison between the two cannot fully explain the corrosion phenomenon. Therefore, the pristine fresh Al was utilized to clarify the passivation, passivity breakdown, and localized corrosion processes during battery operation. Meanwhile, the experimental drawback between RA and FA would be erased mentioned above.
Passivation and corrosion of Al
The electrochemical oxidation window of the LiPF6-based electrolyte is restrained at 4.55 V versus Li/Li+ in the three-electrode cell. Whereas the practical stability of the battery electrolyte would be shrunken due to the volumetric effect and the catalysis of electrode materials11,42. Following the harmful parasitic reactions of the electrolyte would occur at the interface. We detected the current densities and morphology changes at the interface of electrolyte/Al substrate with electrode potentials. Due to the high finish of pristine Al, the surface had no obvious morphological characteristics except from rolling grain (Supplementary Fig. 4). The XRD pattern of Al was revealed in Supplementary Fig. 5 which meant the domination of (220) and (311) orientations (consulting Al PDF#04-0787). The detecting potential chosen is 4.0 V according to the anodic peak (Supplementary Fig. 3) and voltage platform of typical batteries.
Generally, the AFM with Kelvin probe force microscopy (KPFM) module was frequently employed to determine the local topography and corresponding surface potentials (SPs) of various samples. It is sensitive of SPs to the topographic fluctuation and defect distribution function on the surface of these samples and the potential implies the regional electric-field distribution52. Figure 4 displayed that the Al foil after 4 V treatment for 100 h showed a more heterogeneous morphology and SP than those of Al-treated 3rd cycling (Supplementary Figs. 6–9). The latter had a root mean square (RMS) roughness of 38.11 nm, whereas the RMS of the 4 V-treated Al was about 70.52 nm. The difference in RMS values exposed that the prolonged cycling significantly changed the topography of Al, which was coincident with the results of RA. Different from the initial state, new substances accumulated on the surface of polarized Al (Supplementary Fig. 6c–e and Fig. 4i), which verified the outward dissolution of Al3+ and its fluoridation/oxidation (Fig. 4j). A significant enlargement of SP for (4 V-100 h) Al foil in comparison to 3rd cycling Al verified the insulating character of passivation layers Fig. 4d–g. This SP enlargement would prompt the tendency of degrading electronic transfer properties in turn protecting Al substrate from dissolution within a period. The local districts in Fig. 4h, i presented the nonuniform surface of Al foil with the increase in operational time. Eventually, the dilemma of regional overcharge/over-discharge would be encountered in the following Li+ ion insertion and extraction.

AFM of (a) Al foil after 3rd cycle and (b) Al foil after 4 V-100 h treatment. c The height distribution of (a) and (b). d The potential of Al foil after the 3rd cycle. e Potential of Al foil after 4 V-100 h corrosion. f Potential distribution of X (the 3rd cycle). g I. Potential distribution of X, II, Potential distribution of Y (4 V-100 h corrosion). h, i Partial 3D Z sensor of fresh Al after 3rd cycle and fresh Al after 4 V-100 h corrosion. j SEM/EDS of Al foil after 4 V-100 h treatment.
The behavior disclosed that Al entered a passivation state at 4.0 V vs. Li/Li+. However, Al would experience the higher potential accompanying battery charge even overcharge, especially high voltage batteries. The detections of Al oxidation stability at higher potentials were exigent. In Fig. 5, the current densities in Al||Li batteries saturated showing no fluctuations at 4.0, 4.2, and 4.3 V (vs. Li/Li+), while the current densities fluctuated at 4.4, 4.5, 4.6, 4.8, and 5.0 V (vs. Li/Li+). These phenomena presented that the interface behaviors were inhomogeneous at these steps and coupled with electrolyte oxidation. Eventually, the current densities were all saturated by 10 ~ 20 μA cm−2, whose values were fiercer than the literature4,53. The only distinction of these results was the compositions of the electrolytes, which indicated the impacts of solvents/salts mass ratios and species on electrode corrosion. Particularly, the solvating effects on Al3+ and solubility of [AlxMy]− in certain solvents are of vital importance for the oxidation stability of cathodes54.

Chronoamperometry results measured at various applied potentials, a the whole i-t curves for pristine Al||Li at 4, 4.2, 4.3, 4.4, 4.6, 4.8, and 5.0 V. The current collectors were cycled 3 times with a scanning rate of 10 mV s−1. SEM and AFM images for Al foils at (b, c, h) 4.2 V, (d, e, i) 4.4 V, and (f, g, j) 4.8 V.
Nanograins were generated on the surface of Al foil in Fig. 5b–g and Supplementary Fig. 7c–e, which coincident with the results of AFM-KPFM detection. Meanwhile, deposits appeared on the matrix surface of the 4 V-polarized Al substrate, while holes and cracks also appeared on the matrix surface of 4.2, 4.3, 4.4, 4.5, 4.6, 4.8, and 5.0 V-treated Al (Fig. 5b–g and Supplementary Figs. 7e, f, 8 and 9). The holes and cracks demonstrated that the original/newborn layers were incompatible with the substrate, which accumulated tensile stress. In these holes and cracks, stuck particles might be generated because of the inward diffusion of F− (Fig. 6a, b). AlF3 almost disappeared at about 20 nm depth of both 4.4 V and 4.8 V-treated samples (Area ratio: 0.01, Supplementary Fig. 10), which was different from the non-polarized handled sample (Fig. 1f). The phenomenon indicated that external polarized potential arose from the fiercer Al dissolution. The outward diffusion rate of Al3+ was bigger than the inward diffusion rate of F−, and the passivation layer only conserved natural oxides. The Al 2p was detected on the side of the Li anode surface, especially for the 4.8 V-treated sample (Supplementary Fig. 11), which meant the dissolution, penetration, and migration of Al into the electrolyte. However, the Stokes radius of Al3+ may restrict the penetration into a separator and deposited on the Li anode surface, so there was a little trace of Al accumulation29. The dissolved Al ions would reductively deposit on the anode surface, where they break down the SEI structure (Supplementary Fig. 12a–c) and disturb Li+ transfer kinetics due to the alloying effect, leading to irreversible active Li+ loss and impedance growth. Considering the phenomenon, the battery performance deterioration from Al corrosion might be underestimated.

a–c The depiction of Al’s external and internal microstructure changes. d Passivation, passivity breakdown, and corrosion illustration of Al current collector in LIBs with DFT results (e).
Discussion
In a word, the topography differences of Al foils presented that Al foil confront deteriorating risks at the cutoff voltages larger than 4.4 V (Fig. 6a). The passivation layers still existed until the electrode potentials were applied at 4.2 V. During this period, the natural oxide layer was fluoridized gradually deeper than 20 nm. Nanograins (AlF3/Al2O3) were accumulated on the surface of 4.0 V-treated Al, whose Al3+ was from the original oxide film. Beyond 4.2 V (<4.6 V), the inward diffusion of F− across the grain boundaries resulted in the fluoride generation on the Al internal substrate, which provided cracks and crevices. Upon 4.6 V, the surface film came porous, stripped, and cracked with some protuberances, which meant the sharp outward diffusion of Al3+. The thickness of the surface fluoride film was shortened to less than 20 nm. When the potential was higher than 4.8 V, the original oxide and fluoride layers almost disappeared with a little trace, and the newborn layers were immediately generated on bare Al. The subsequent passivation behavior on the high voltages meant the unremitting consumption and contamination of the electrolyte. Importantly, oxygen loss of cathode materials would dominantly proceed with the electrolyte oxidation rather than electrochemical oxidation at high voltage, which meant the catalytic effect of active materials to parasitic reactions of the electrolytes. The electrolyte decomposition always existed in the whole process, which was evidenced by the fluorides of Al. So, the corrosion of the Al substrate would be strengthened with active material loading. The nature of passivation layers varies with the applied potential states, and it is urgent to develop new passivating layers on current collectors for higher energy requirements.
During battery operation, dissolution/passivation breakdown/corrosion of Al and cathode was inevitable in Fig. 6 and Supplementary Fig. 13. Al current collectors underwent the inward diffusion of F− and outward dissolution of Al3+, which was depicted in Fig. 6b, c. Combining Fig. 1f, there was a foreign passivation layer accumulated on the surface of Al2O3/Al (Supplementary Fig. 10), which was mainly composed of Al-O-F and AlF3 (Fig. 6d, Passivation reaction 1–2). The phenomenon indicated the localized transformation of Al2O3 to fluorides of the substrate (Eqs. (1)–(4)), which came with the external and internal microstructure changes in Fig. 6a, b. So, the above-mentioned tensile stress arose. Additionally, the expansion of microstructural volume could be expressed at about 64% according to Eq. (4), which leaded to cracks:
The presence of AlF3 would introduce Al3+ vacancy into the original transitional-passivating layer and destroy its ordered atomic packing structure55. The formation of Al3+ vacancy would accelerate ion diffusion, which means fast electrolyte reactions, especially in crevices. Active materials’ loading is more likely to squeeze the electrolyte with one-dimensional wormhole contact on Al foil, which leads to a localized concentration of corrosive media.
On the other hand, the decomposition of the electrolytes accompanied not only the generation of electrolyte interfaces but also the replacement of Al2O3 to AlF3 and ongoing corrosion of the Al substrate (Fig. 6c). The latter phenomena would regulate the anodic stability of Al despite the porous native oxide film on the Al substrate failing to protect Al during battery charging/discharging. Binding energy calculations were implemented to clarify the insertion/adsorption mechanisms of HF/EC into the Al substrate. As seen in Fig. 6c, the cleaving surface of the substrate chose Al (111), (220), and (311). The results elucidated that the most prior adhesion of uncoordinated polar EC molecules covers the surfaces of Al, especially (220) whose binding energy is much higher than that of the others. Thus, the relatively stronger interaction between polarized Al substrate and EC molecules is more possibly to induce corrosion bridge/couple. Even though the highest occupied molecular orbital (HOMO) energy level indicates that the DEC has a much higher HOMO value than that of EC56. Moreover, EC has a stronger coordination ability with HF than DEC, which means EC will coerce HF to penetrate Al crevices at the charging state57. Consequently, the more intensive attraction toward the Al substrate not only accelerates the EC electrochemical oxidation reaction but also extensively changes the composition of electrolytes. This insight sheds light on the solvent design for higher voltage application, which can be summarized as the weak interaction of selected polar solvents helping to achieve a stable interfacial model and fast kinetics of electrolyte carrier ions.
In summary, Al current collectors suffer from corrosion during battery operation. At the end of battery life, the Al current collector presented uneven passivation and passivity breakdown. The structural destruction of passivity breakdown and localized corrosion enhanced the electrochemical activity of the reclaimed Al substrate, which resulted in the multiplied anodic current densities. This phenomenon was verified by the lower CE (1.14% loss) in a LiCoO2||Li half-cell using RA than that using FA, which means the irreversible capacity was partially contributed by the passivity breakdown of the Al current collector. Over the entire battery life, the cycle/rate performances of LiCoO2||Li half cells degraded with capacity loss >20% because of the corrosion of the Al current collector. The inward diffusion of F− was sustained till 4.4 V polarized state, while the outward diffusion of Al3+ took the lead in the corrosion process when the applied charge voltage was larger than 4.4 V. Therefore, more attention should be paid to engineering the Al current collectors to suppress their corrosion, which is an important part of making long-lasting and reliable LIBs. Also, the detection of Al corrosion in LIBs is pro-found to the exploration of high-energy-density batteries, which use Al as a cathode current collector.
Methods
Materials
LiCoO2, pristine fresh Al foil, high-purity Li foil, and the solvents were purchased from Guangdong Canrd New Energy Technology Co., Ltd (the parameters shown in Supplementary Table 1). The electrolyte was prepared by solving 1 M LiPF6 in an EC/DEC/FEC mixture with a volume ratio of 3:6:1. The preparation of the electrolyte was carried out in an argon-filled glovebox with an H2O concentration of <0.1 ppm and an O2 concentration of <0.1 ppm. The contents of H2O of the solvents (EC/DEC/FEC) were all below 20 ppm. The water content of LiPF6 conductive salt was below 50 ppm, and the free acid HF was restrained within 100 ppm. Waste LiCoO2 batteries (Huizhou Desay Battery Co., Ltd. Model: 18S2001-AL, limited charge voltage: 4.3 V) were discharged and disassembled to obtain reclaimed Al foil for performance detection. Before the battery was wasted, they experienced a discharge process at room temperature, and the process continued until the battery capacity decayed to less than 50% of the rated capacity. After the complete charge/discharge process, the status of reclaimed Al was the stage after electrode corrosion occurrence assured by the same operation of all batteries. The reclaimed Al from LiCoO2 batteries (18S2001-AL) was named RA.
Both the RA from LiCoO2 batteries and fresh Al (FA) foils were rinsed with DEC and then N-methyl-2-pyrrolidone (NMP) to remove surface contaminants in an argon-filled glovebox. The above Al foils were applied as cathode current collectors after the microscopy detection shown in Supplementary Fig. 1. For cathode materials, LiCoO2, polyvinylidene fluoride (PVDF), and acetylene black were mixed and ground at a mass ratio of 8:1:1, and NMP was added to prepare the slurry. The slurry was evenly stirred and coated on the surface of different Al foils, where the loading of the active material was ~2.3 mg cm−2. The electrode samples were sliced after drying at 80 °C for 12 h, and 2032-type coin cells were assembled. The dosage of the electrolyte was 30 μl for all coin cells. Also, different Al foils were punched to 1.13 cm2 before transferring to the Ar glovebox for Al||Li coin-cell assembly and electrochemical experiments.
Electrochemical
LiCoO2 on different Al foils was employed to determine the cathode performance degradation from the corrosion of current collectors. The current densities of LiCoO2||Li cells were set to 1 C = 200 mAh g−1, respectively. Cycle performances were carried out at 0.5, 1, and 5 C with the ambient temperature. The Al||Li half-cells were utilized to detect the interfacial corrosive reactions of the electrolyte with Al current collectors. Cyclic voltammetry (CV) was used to determine the electrochemical behaviors of the Al foils before and after the battery charge/discharge process, and the potential range was 2–6 V with a scanning speed of 10 mV s−1 at room temperature. The EIS of RA/FA||Li half-cells in the electrolyte (1 M LiPF6 in EC/DEC/FEC) were tested on CHI 760E with an amplitude of 5 mV. The frequency ranged from 0.1 Hz to 105 Hz at the initial potentials of open circuit potential (OCP) and 4.6 V vs. Li/Li+. Equivalent circuits of EIS were fitted by Zview 3.1.
The CV and EIS tests in a 3-electrode configuration were performed with the CR-2032 coin-type half cells in the atmosphere, which were different from the traditional open electrochemical system. The Al foil was assembled as the working electrode, and the Li foil was used as reference and counter electrodes. All assembling work was arranged in an argon-filled glovebox, with the content of O2 and H2O maintained below 0.1 ppm.
The pristine fresh Al foil was applied to determine the corrosion mechanisms in LIBs. Considering the voltage platform of insertion/extraction of Li+ in LiCoO2, the chronoamperometry experiment of FA foil was carried out at the potential of 4 V. Additionally, once the coin-cells of FA||Li were fabricated, they were held at 2–6 V for 3 cycles before chronoamperometry tests (at 4.0, 4.2, 4.3, 4.4, 4.5, 4.6, 4.8, and 5.0 V vs. Li/Li+ for 24 h). This measure can minimize the errors from incomplete passivation of the Al and more closely simulate the situations where the cathode electrolyte interface (CEI) is stabilized.
Characterization and theory calculation
The RA/FA samples after electrochemical tests were disassembled and washed using flowing DEC in the glove box. Then, they were left to dry overnight in the Ar glove box for further surface characterization. Field-emission scanning electron microscopy (SEM, ZEISS Gemini 300, Inlens and ET secondary electron detectors), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha; Vacuum degree of analysis room: ~3 × 10−7 mba; Operating voltage: 12 kV; Beam spot: 400 μm) were conducted on the surface of FA and RA foils for the detection of the morphologies and composition of the current collectors’ sides facing the Li anode. For XPS, the X-ray source adopted an Al Kα (1486.6 eV). Surface contamination C1s (284.8 eV) was used as the standard for correction. Full spectrum scanning energy was 150 eV, and the step size was 1 eV. Narrow-spectrum scanning had a pass energy of 50 eV and a step size of 0.1 eV. The Al foils were etched by a beam of Ar+ to achieve the high-resolution determination from the surface to the vertical inner regions of the foils. The surfaces of Li anodes were also detected by XPS. Atomic force microscopy (AFM, Bruker Dimension Icon (Germany), tapping contact) accompanied by Kelvin probe force microscopy (KPFM) was used to attain the topography and corresponding surface potential of the samples before and after passivation/corrosion. The scanning range of samples was 10 × 10 × 10 μm, and the resolution was 0.04 nm in the vertical direction and 0.15 nm in the lateral direction. The roughness of all Al foils was detected by AFM.
The binding energies of polar molecules HF and EC with Al were calculated. All geometry optimizations and energy calculations were implemented by DFT (the Perdew-Burke-Ernzerhof (PBE) functional in the generalized gradient approximation (GGA))58. For the calculations of the binding energies of one HF/EC molecule adsorbed on the surface of Al, the 5 × 5 supercells of Al(111), (220), and (311) slabs with five layers were built corresponding to the theoretical equilibrium lattice constant. The positions at the bottom two layers of the Al slabs were fixed as the Al bulk position, and the vacuum length was set at 15 Å. The kinetic cutoff energy 520 eV was applied, and the Al lattice parameters were optimized employing a Monkhorst-Pack grid k-point of 4 × 4 × 4. All the atoms were optimized until the total energies converged to below 10−4 eV and the forces acting on atoms were less than 10−2 eV Å−1. The binding energies of the adsorbed molecule are defined as:
where EM/Al and EAl are the energies of the Al slab (i.e., (111)/(220)/(311)) with and without an M molecule (M represents HF/EC molecule). EM is the energy of an M molecule in the vacuum.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Zhong, S. et al. Recent progress in thin separators for upgraded lithium ion batteries. Energy Storage Mater. 41, 805–841 (2021).
Shi, J. et al. Composite membrane with ultra-thin ion exchangeable functional layer: a new separator choice for manganese-based cathode material in lithium ion batteries. J. Mater. Chem. A 3, 7006–7013 (2015).
Choudhury, R., Wild, J. & Yang, Y. Engineering current collectors for batteries with high specific energy. Joule 5, 1301–1305 (2021).
Yoon, E., Lee, J., Byun, S., Kim, D. & Yoon, T. Passivation failure of Al current collector in LiPF6-based electrolytes for lithium-ion batteries. Adv. Funct. Mater. 32, 1–10 (2022).
Wang, M. et al. Graphene-armored aluminum foil with enhanced anticorrosion performance as current collectors for lithium-ion battery. Adv. Mater. 29, 1–7 (2017).
Chen, J. et al. Dynamic intelligent Cu current collectors for ultrastable lithium metal anodes. Nano Lett. 20, 3403–3410 (2020).
Fu, A. et al. Lithiophilic and antioxidative copper current collectors for highly stable lithium metal batteries. Adv. Funct. Mater. 31, 1–9 (2021).
Wen, Z. et al. Anticorrosive copper current collector passivated by self-assembled porous membrane for highly stable lithium metal batteries. Adv. Funct. Mater. 31, 1–11 (2021).
Tebbe, J. L., Fuerst, T. F. & Musgrave, C. B. Degradation of ethylene carbonate electrolytes of lithium ion batteries via ring opening activated by LiCoO2 cathode surfaces and electrolyte species. ACS Appl. Mater. Interfaces 8, 26664–26674 (2016).
Flamme, B. et al. Guidelines to design organic electrolytes for lithium-ion batteries: environmental impact, physicochemical and electrochemical properties. Green Chem. 19, 1828–1849 (2017).
Cao, Z. et al. Electrolyte solvation engineering toward high-rate and low-temperature silicon-based batteries. ACS Energy Lett. 7, 3581–3592 (2022).
Li, M., Wang, C., Chen, Z., Xu, K. & Lu, J. New concepts in electrolytes. Chem. Rev. 120, 6783–6819 (2020).
Qi, S., Tang, X., He, J., Liu, J. & Ma, J. Construction of localized high-concentration PF6− region for suppressing NCM622 cathode failure at high voltage. Small Methods 2201693, 1–7 (2023).
Qian, K. et al. Decoupling the degradation factors of Ni-rich NMC/Li metal batteries using concentrated electrolytes. Energy Storage Mater. 41, 222–229 (2021).
Yin, S. et al. Chemical-mechanical effects in Ni-rich cathode materials. Chem. Mater. 34, 1509–1523 (2022).
Jiang, M., Danilov, D. L., Eichel, R. A. & Notten, P. H. L. A review of degradation mechanisms and recent achievements for Ni-rich cathode-based Li-ion batteries. Adv. Energy Mater. 11, 2103005 (2021).
Hou, J. B. et al. Modified tungsten oxide as a binder-free anode in lithium-ion battery for improving electrochemical stability. Tungsten 4, 356–369 (2022).
Miao, S. C. et al. Transition metals for stabilizing lithium metal anode: advances and perspectives. Tungsten https://doi.org/10.1007/s42864-023-00231-3 (2023).
Pender, J. P. et al. Electrode degradation in lithium-ion batteries. ACS Nano 14, 1243–1295 (2020).
Gabryelczyk, A., Ivanov, S., Bund, A. & Lota, G. Corrosion of aluminium current collector in lithium-ion batteries: a review. J. Energy Storage 43, 103226 (2021).
Du, P. et al. Research progress towards the corrosion and protection of electrodes in energy-storage batteries. Energy Storage Mater. 57, 371–399 (2023).
McOwen, D. W. et al. Concentrated electrolytes: decrypting electrolyte properties and reassessing Al corrosion mechanisms. Energy Environ. Sci. 7, 416–426 (2014).
Guo, L., Thornton, D. B., Koronfel, M. A., Stephens, I. E. L. & Ryan, M. P. Degradation in lithium-ion battery current collectors. J. Phys. Energy 3, 031501 (2021).
Kolesnikov, A. et al. Galvanic corrosion of lithium-powder-based electrodes. Adv. Energy Mater. 10, 1–9 (2020).
Peng, J. et al. Surface coordination layer passivates oxidation of copper. Nature 586, 390–394 (2020).
Xu, X. et al. Greatly enhanced anticorrosion of Cu by commensurate graphene coating. Adv. Mater. 30, 1702944 (2018).
He, X. et al. The passivity of lithium electrodes in liquid electrolytes for secondary batteries. Nat. Rev. Mater. 6, 1036–1052 (2021).
Delmas, C. Sodium and sodium-ion batteries: 50 years of research. Adv. Energy Mater. 8, 1–9 (2018).
Hosaka, T., Kubota, K., Hameed, A. S. & Komaba, S. Research development on K-ion batteries. Chem. Rev. 120, 6358–6466 (2020).
Lee, W. & Park, S. J. Porous anodic aluminum oxide: anodization and templated synthesis of functional nanostructures. Chem. Rev. 114, 7487–7556 (2014).
Zhu, P. et al. A review of current collectors for lithium-ion batteries. J. Power Sources 485, 229321 (2021).
Chidiac, J., Timperman, L. & Anouti, M. Small dissymmetry, yet large effects on the transport properties of electrolytes based on imide salts: consequences on performance in Li-ion batteries. J. Energy Chem. 65, 352–366 (2021).
Cho, E. et al. Corrosion/passivation of aluminum current collector in bis(fluorosulfonyl) imide-based ionic liquid for lithium-ion batteries. Electrochem. Commun. 22, 1–3 (2012).
Kramer, E., Passerini, S. & Winter, M. Dependency of aluminum collector corrosion in lithium ion batteries on the electrolyte solvent. ECS Electrochem. Lett. 1, 9–12 (2012).
Abouimrane, A., Ding, J. & Davidson, I. J. Liquid electrolyte based on lithium bis-fluorosulfonyl imide salt: aluminum corrosion studies and lithium ion battery investigations. J. Power Sources 189, 693–696 (2009).
Benbouzid, A. Z. et al. Ionic liquid route for the corrosion inhibition of Al alloys: the effect of butylammonium nitrate on the corrosion of AA2024-T6. Corros. Commun. 9, 57–64 (2023).
Umesh, B. et al. High-Li+-fraction ether-side-chain pyrrolidinium–asymmetric imide ionic liquid electrolyte for high-energy-density Si//Ni-rich layered oxide Li-ion batteries. Chem. Eng. J. 430, 132693 (2022).
Krämer, E. et al. Mechanism of anodic dissolution of the aluminum current collector in 1 M LiTFSI EC:DEC 3:7 in rechargeable lithium batteries. J. Electrochem. Soc. 160, A356–A360 (2013).
Yang, S. et al. MXene-Ti3C2 armored layer for aluminum current collector enable stable high-voltage lithium-ion battery. Adv. Mater. Interfaces 9, 2200856 (2022).
Lu, J. et al. Effectively suppressing dissolution of manganese from spinel lithium manganate via a nanoscale surface-doping approach. Nat. Commun. 5, 1–8 (2014).
Wandt, J. et al. Transition metal dissolution and deposition in Li-ion batteries investigated by operando X-ray absorption spectroscopy. J. Mater. Chem. A 4, 18300–18305 (2016).
Rinkel, B. L. D., Hall, D. S., Temprano, I. & Grey, C. P. Electrolyte oxidation pathways in lithium-ion batteries. J. Am. Chem. Soc. 142, 15058–15074 (2020).
Ma, D. et al. An all-weather Li/LiV2(PO4)3 primary battery with improved shelf-life based on the: in situ modification of the cathode/electrolyte interface. J. Mater. Chem. A 8, 16951–16959 (2020).
Cong, L. et al. Role of perfluoropolyether-based electrolytes in lithium metal batteries: implication for suppressed Al current collector corrosion and the stability of Li metal/electrolytes interfaces. J. Power Sources 380, 115–125 (2018).
Zhu, P. et al. Direct reuse of aluminium and copper current collectors from spent lithium-ion batteries. Green Chem. https://doi.org/10.1039/d2gc03940k (2022).
Zheng, Q. et al. Compact 3D metal collectors enabled by roll-to-roll nanoimprinting for improving capacitive energy storage. Small Methods 6, 1–13 (2022).
Zhang, H., Yu, X. & Braun, P. V. Three-dimensional bicontinuous ultrafast-charge and-discharge bulk battery electrodes. Nat. Nanotechnol. 6, 277–281 (2011).
Naumkin, A. V., Kraut-Vass, A., Gaarenstroom, S. W. & Powell, C. J. NIST X-ray photoelectron spectroscopy database. https://srdata.nist.gov/xps/selEnergyType.aspx, https://doi.org/10.18434/T4T88K (2012).
Hess, A., Kemnitz, E., Lippitz, A., Unger, W. E. S. & Menz, D. H. ESCA, XRD, and IR Characterization of Aluminum Oxide, Hydroxyfluoride, and Fluoride Surfaces in Correlation with Their Catalytic Activity in Heterogeneous Halogen Exchange Reactions. J. Catal. 148, 270–280 (1994).
Qu, D. et al. Electrochemical impedance and its applications in energy-storage systems. Small Methods 2, 1–27 (2018).
Vivier, V. & Orazem, M. E. Impedance analysis of electrochemical systems. Chem. Rev. 122, 11131–11168 (2022).
Adhitama, E. et al. Revealing the role, mechanism, and impact of AlF3 coatings on the interphase of silicon thin film anodes. Adv. Energy Mater. 12, 2201859 (2022).
Li, X., Chen, Y., Nguyen, C. C., Nie, M. & Lucht, B. L. Stability of inactive components of cathode laminates for lithium-ion batteries at high potential. J. Electrochem. Soc. 161, A576–A582 (2014).
Wang, X., Yasukawa, E. & Mori, S. Inhibition of anodic corrosion of aluminum cathode current collector on recharging in lithium imide electrolytes. Electrochim. Acta 45, 2677–2684 (2000).
Kim, J. W. et al. Surface chemistry of LiNi0.5Mn1.5O4 particles coated by Al2O3 using atomic layer deposition for lithium-ion batteries. J. Power Sources 274, 1254–1262 (2015).
Zhang, W., Yang, T., Liao, X., Song, Y. & Zhao, Y. All-fluorinated electrolyte directly tuned Li+ solvation sheath enabling high-quality passivated interfaces for robust Li metal battery under high voltage operation. Energy Storage Mater. 57, 249–259 (2023).
Ye, C. et al. Converting detrimental HF in electrolytes into a highly fluorinated interphase on cathodes. J. Mater. Chem. A 6, 17642–17652 (2018).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
We greatly thank the financial support from the Fundamental Research Funds for the Central Universities (2042023kf0214) and the 111 Project (B16009).
Author information
Authors and Affiliations
Contributions
Pin Du: conceptualization, methodology, investigation, writing—original draft, writing—review & editing, visualization. Jiale Wan: writing—review & editing. Jiakang Qu: writing—review & editing. Hongwei Xie: supervision. Dihua Wang: supervision. Huayi Yin: project administration, conceptualization, writing—review & editing, formal analysis, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, P., Wan, J., Qu, J. et al. Passivation and corrosion of Al current collectors in lithium-ion batteries. npj Mater Degrad 8, 43 (2024). https://doi.org/10.1038/s41529-024-00453-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-024-00453-x
