
Abstract
Pseudomonas aeruginosa is a major opportunistic pathogen responsible for severe infections in immunocompromised patients. The contamination of drinking water networks (DWNs) with this pathogen is underestimated, as it is mostly in the state of persister cells undetected by the recommended monitoring technique. We collected water samples from eight cities distant from each other and searched for P. aeruginosa using a culture-based method that resuscitates persister cells. The genomes of isolates were sequenced. Five DWNs of the eight tested (62.5%) were contaminated with P. aeruginosa, of which four were contaminated with high-risk clones (ST308, ST395). Surprisingly, the ST308 isolates retrieved from the four independent and distant DWNs were clonal. Most P. aeruginosa isolates shared a genomic island conferring tolerance to copper-ions. The population structure of the collection may result from both a common source of contamination by plumbing supplies and the selection of clones sharing genetic elements that presumably aided their propagation in DWNs.
Similar content being viewed by others

Introduction
Pseudomonas aeruginosa is an opportunistic pathogen responsible for severe infections in immunocompromised patients1,2,3. This species is ubiquitous in soil, water, and other moist environments, including drinking water networks (DWNs) and those in hospitals. P. aeruginosa often contaminates the distal parts of water distribution systems, such as taps, sinks, showers, and U-bends, also called sink traps2,4,5, and can thus contaminate patients through water, leading to waterborne nosocomial infections6,7,8. It is responsible for a broad range of diseases, including respiratory infections in mechanically ventilated patients, bloodstream infections, and infections of wounds from burn injuries, which can lead to death9. Outside of hospitals, tap water is a source of contamination by P. aeruginosa for nebulizers used by cystic-fibrosis patients10.
European regulations require monitoring for P. aeruginosa in DWNs by immediate filtration and culture on agar media11,12. However, this method does not detect persister cells13. Indeed, water distribution systems are hostile environments for bacterial development, with nutrient-poor conditions and the presence of inhibitors (copper ions, chlorine). This favors the development of persister cells, also called viable but not-culturable, that are no longer culturable on media upon which they are usually able to grow14,15. Although not culturable, these cells can be resuscitated and recover their full virulence under suitable conditions16.
Using a culture-based method that resuscitates persister cells, we previously found that a copper-tolerant high-risk clone of P. aeruginosa ST308 contaminated the DWN of a University Hospital in France5. This clone was tolerant to copper due to a genomic island (called GI-7) that probably originated from environmental Pseudomonas spp5.
The source of contamination of DWNs with P. aeruginosa is still unclear. This pathogen can contaminate drinking water reservoirs17 but water treatment is usually efficient in removing it before the water enters DWNs18. By contrast, P. aeruginosa has been repeatedly isolated from water meters prior to their installation19.
Whether the presence of a high-risk clone in the plumbing system of such premises was an isolated event or a concern for other DWNs is unknown. Here, we searched for P. aeruginosa in eight independent and distant DWNs in France, including both healthcare and community DWNs. We determined the population structure of the bacterial isolates using genome-based typing, allowing the identification of genetic elements shared by P. aeruginosa that contaminate DWNs and providing clues about the source of contamination.
Results and discussion
Contamination of drinking water networks by P. aeruginosa in France
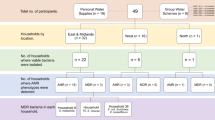
Using a method that resuscitates persister cells, we previously detected high-risk clones of P. aeruginosa in the DWNs of a hospital and a long-term care facility, both located in Besançon (France), in 2017–20185,20 (Table 1, Fig. 1). We tested the extent of such contamination throughout the country by broadening our sampling to seven other French cities and retesting the DWNs of Besançon. Among the collected water samples, 5% (12/239) were positive for P. aeruginosa. We found P. aeruginosa in the DWNs of five of the eight cities tested. This pathogen contaminated three of the eight hospital DWNs and two of the five community networks tested (Table 1). All isolates of P. aeruginosa were susceptible to all antibiotics tested. As culture-based methods may fail to detect persister cells of P. aeruginosa in water5, our findings highlight the urgency for new recommendations for the monitoring of drinking water contamination that consider the presence of persister cells.

DWNs, drinking water networks.
Most of the P. aeruginosa isolates detected in the drinking water networks belong to high-risk clones
The sequence type (ST) of all 12 isolates were first determined by PCR and sequencing. Five isolates belonged to the high-risk clone ST308, three to the high-risk clone ST395, two to ST27, one to ST606, and one to ST3299 (Table 1, Fig. 1). We then sequenced the genome of one isolate representative of each ST per sample site for 10 isolates in total. The DWNs of four cities (Besançon, Bordeaux, Nantes, and Nîmes) were contaminated with the high-risk clones ST308 or ST395. ST308 was found in the community networks of Bordeaux and in the hospital networks of Nantes and Nîmes (Table 1). Of note, ST308 was also retrieved during the 2017 sampling campaign in the hospital network of Besançon5 (Table 1). ST308 has been responsible for outbreaks in hospitals where water was reported as a potential reservoir of P. aeruginosa6,8. We also retrieved ST395 isolates from the community DWN of Besançon (Table 1). This ST was responsible for a large hospital outbreak in the same city, as well as in the United Kingdom, where the authors suggested that P. aeruginosa ST395 rapidly colonized the plumbing system of a new hospital, before its commissioning and was transmitted to patients by the water7,21. Although ST27 and ST606 are not yet classified as high-risk clones, they have emerged in European countries and can produce metallo-ß-lactamases of Ambler class B (VIM-2 and VIM-4 for ST27, and IMP-15 for ST606)22,23.
The biological features needed to survive in the DWNs (resistance to inhibitors and predators) suggest that P. aeruginosa STs contaminating DWNs may have gathered in a clade. We tested this possibility by localizing these STs on the phylogenetic tree of all currently known STs of P. aeruginosa (Fig. 2a) and found the six STs (Table 1) to be scattered throughout the tree, suggesting no evidence of clades specifically adapted to this niche.

a shows the distribution of the STs of the Pseudomonas aeruginosa strains isolated from hospital and community drinking water networks (blue squares) on an unrooted tree built using the data of all known STs of P. aeruginosa (n = 753) and cgMLST data (3786 genes). The scale bar indicates the genetic distance in number of substitutions per site. The green squares represent the ST of reference strains PAO1, PA7, and PA14. Strain PA7 now belongs to the species Pseudomonas paraeruginosa and is no longer classified as P. aeruginosa39. Scale bar refers to a phylogenetic distance of 0.01 nucleotide substitutions per site. b is the distribution of Pseudomonas aeruginosa ST308 isolated in hospital and community drinking water networks on a phylogenetic tree built from all 95 genomes of ST308 isolates deposited at the NCBI by April 5, 2021, based on the wgMLST of ST308 (2937 genes). The scale is the length of branch that represents 0.00001 nucleotide substitutions per site. The color of the outer circle indicates the origin of the isolates (red for human, blue for environment, green for unknown origin). c is the minimum spanning tree (built from wgMLST distances) that grouped the P. aeruginosa ST308 strains isolated from the drinking water networks in France.
Seeking a common source of P. aeruginosa in drinking water networks
ST308 was the most highly represented ST (4 of 10 isolates) in the 2019–2020 campaign and was also repeatedly found in the DWN of the hospital of Besançon in 2017 (Table 1, Fig. 1). Understanding the population structure of ST308 in French DWNs could help to identify the source of contamination. We compared the genomes of the ST308 isolates found in the DWNs of Nantes (wPA-Nan1), Nîmes (wPA-Nim2), Besançon (wPA-Bes1, wPA-Bes4, wPA-Bes12, for which the genomes were available from a previous study5), and Bordeaux (wPA-Bor1) with those of the 95 ST308 strains for which the genomes were deposited at the NCBI. Surprisingly, we found that isolates from the French DWNs grouped together on the phylogenetic tree of ST308 (Fig. 2b), with less than nine genes with different alleles (Fig. 2c).
Such genetic proximity between ST308 isolates collected in 2019–2020 in distant DWNs was unexpected and raised suspicions. Two factors allowed us to rule out cross-contamination at the laboratory. First, water samples positive for P. aeruginosa ST308 were processed at different times by the Infection Control laboratory of the UHB (Table 1). Second, given the mutation rate of P. aeruginosa (~10–15 SNPs per year), the number of genomic differences between these ST308 isolates was higher than that between the genomes of contaminating strains in the laboratory24.
This clonal distribution strongly suggests a common source of contamination with P. aeruginosa ST308 in the DWNs of four distant cities in France (Fig. 3). As the DWNs of these four cities are supplied by independent water sources, it is reasonable to exclude them as a reservoir of P. aeruginosa ST308. However, DWNs are built with supplies, such as water meters, provided by a small number of manufacturers, which calibrate water meters on test benches before shipment to the clients. The calibration of water meters has been identified as a high-risk activity for contamination by P. aeruginosa. Hence, a technical report in 2016 indicated that 23% of the brand-new water meters from the stock of water supply companies were contaminated with P. aeruginosa19. It is thus possible that water meters could inoculate a DWN with P. aeruginosa that further disseminates and persists for years in the network. This hypothesis needs to be tested by additional studies. However, collections of P. aeruginosa isolated from water meters prior to their installation are not available.

The number of samples collected from hospital and community drinking water networks is indicated, along with the sampling period. The water samples came from the networks of the cities of Amiens (49.875 N, 2.253 E), Angers (47.484 N, 0.556 W), Besançon (47.225 N, 5.963 E), Bordeaux (44.828 N, 0.604 W), Nantes (47.211 N, 1.554 W), Nîmes (43.825 N, 4.321 E), Tours (47.387 N, 0.668 E), and Trévenans (47.577 N, 6.872 E).
Drinking water network: niche speciation of P. aeruginosa persister cells and a source of contamination for patients?
Previous studies reported that the core genomes of ST308 and ST395 harbor the 37-kb genomic island GI-7, which contains an array of 13 genes encoding copper transporters and helps P. aeruginosa to tolerate copper in solution5,21. We confirmed the presence of GI-7 in 24 of the 28 isolates (86%) of our collection contaminating DWNs. GI-7-containing isolates all belonged to ST308, ST309, and ST395. However, the tolerance to copper-ions was not fully specific to GI-721. Although scattered throughout the phylogenetic tree of the species (Fig. 2a), these STs share a biological feature (tolerance to copper-ions) that presumably aided their proliferation and propagation in DWNs.
The few clones of P. aeruginosa surviving and persisting in the DWNs must have been able to cope with the harsh environment of this niche (e.g., nutrient limitation, predation by free-living ameba, and inhibition by copper-ions and chlorine)13. The biocidal properties of copper are exploited for the distribution of water and copper-containing materials are widely used for pipes and the fixtures of plumbing systems, leading to the release of copper-ions into drinking water25. The observed speciation may have resulted from the selection of a few niche specialists (ST308, ST309, ST395) that evolved to overcome the harsh conditions and adapt to the DWNs. We were unable to test this possibility, as any attempt to correlate the presence of copper-resistant P. aeruginosa with the network material was precluded because all the drinking water systems of the participating hospitals were composite, with portions in copper and others in polyvinyl chloride, galvanized steel, or cross-linked polyethylene.
No outbreaks with these high-risk clones of P. aeruginosa were reported during the study period in the tested hospitals. However, UHB experienced an outbreak of P. aeruginosa ST395 between 1997 and 2008, probably originating from the contamination of distal parts of the water distribution system by the drinking water21. Notwithstanding, wgMLST analysis showed that the two isolates of P. aeruginosa ST395 (wPA-Bes15 and wPA-Bes16; Table 1) in the DWN were not clonal with the ST395 clinical isolates coming from the hospitalized patients (data not shown). However, hospital outbreaks with high-risk P. aeruginosa ST308 and ST395 have been repeatedly reported7,8,21,26, with epidemiological investigations, when performed, identifying the DWN as a probable source of contamination7,21. In these studies, P. aeruginosa was cultured from water samples using the recommended method unsuitable for persister cell recovery. Although not immediately culturable, persister cells can resuscitate and recover their full virulence and cultivability under suitable conditions16,25. Persister cells of P. aeruginosa contaminating the DWNs could then act as a reservoir for patient contamination by contaminating and thriving in the distal portion of hospital water distribution systems and then recovering their virulence under more favorable conditions.
Limitations and strengths of the study
Although numerous (n = 239), the water samples were distributed within the DWNs of only eight cities in a single country. Moreover, the number of water samples differed between the DWNs tested (Table 1). Further studies should globally test the presence of persister cells of P. aeruginosa in DWNs. Water sampling during different seasons was another limitation, as water temperature can affect the presence of P. aeruginosa in DWNs, with higher quantities of P. aeruginosa being reported in the spring27. We did not measure the temperature of the water at the time of water sampling. However, low temperature possibly accounted for the absence of P. aeruginosa ST308 in DWNs sampled in the winter (i.e., Tours, Trévenans). Although artificial selection of the specific strains by the method we used for bacterial resuscitation cannot be excluded25, the major strength of this study was the culture-based method that allowed the resuscitation of persister cells, necessary for genome sequencing and comparison at the nucleotide level. A culture-independent typing method (named high-throughput short sequence typing—HiSST) has been recently developed and used to type uncultured environmental Serratia marcescens28. Its extension to P. aeruginosa would facilitate the detection of high-risk clones of this pathogen in DWNs. We also ensured that all water samples and all bacterial isolates were processed in a central laboratory to limit technical deviation.
Here, we searched for P. aeruginosa in the DWNs of eight French cities using a culture-based method that resuscitates persister cells and found five of the networks to be positive. High-risk clones (ST308, ST395) contaminated four of the eight DWNs tested. The close genetic proximity of the ST308 isolates retrieved from four independent and distant DWNs suggests the plumbing supplies as the common source of contamination. However, most of the isolates contaminating the DWNs shared a genomic island that confers tolerance to copper-ions. Overall, this suggests that P. aeruginosa isolates retrieved from DWNs result from both a common source of contamination and the selection of clones sharing genetic elements that presumably aid their proliferation and propagation in DWNs. More attention should be given to persister cells of P. aeruginosa of DWNs, which could contaminate patients after proliferation in water points of use.
Methods
Setting
Between January 2019 and November 2020, we collected 239 water samples from the DWNs of eight French cities (Table 1), for which the geographic location is indicated in Fig. 3. The hospital DWNs in the eight cities were sampled, as well as the community DWNs of four cities (Fig. 3).
Water sampling
One liter of cold-water samples was collected monthly over 3 months for the hospital and community DWNs (Table 1). For the hospitals, we sampled faucets distant from any medical activity to avoid any contamination with P. aeruginosa isolates originating from patients or the environment of care units. Faucets were in technical areas, such as medical equipment storage areas or technical facilities. For each hospital DWN, we sampled the main water inlet and three to five water points of use from the building served by this supply (Table 1). We followed the EN ISO 19458 guidelines for water sampling. Briefly, water was sampled in sterile polypropylene bottles after aerator removal, disinfection of the outlet with a gas burner or a swipe soaked with the disinfectant Aniosurf (ANIOS, Lille-Hellemmes, France), and a 1-min pre-flush29,30. The bottles contained 20 mg.L−1 sodium thiosulphate (VWR, Fontenay-sous-Bois, France) and 100 µM of the copper-ion chelating agent diethyldithiocarbamate (DDTC; Sigma-Aldrich, Saint-Quentin Fallavier, France), which can resuscitate copper-impregnated bacterial cells25. Bottled sterile water (Fresenius Kabi, Sevres, France) was used as a negative control.
Microbiological analysis of the water
All samples were sent to (within 24 h and at room temperature) and analyzed by the Infection Control laboratory of the University Hospital of Besançon (UHB). Samples were stored for 14 days at 22 °C in the dark to promote the resuscitation of copper-stressed P. aeruginosa induced by the chelator DDTC5,25. Then, we filtered three subsamples of 250 mL through 0.45-µm membranes which were then placed on (i) R2A agar (Biokar Diagnostics, Allonne, France) and further incubated for 7 days at 22 °C, (ii) cetrimide-containing agar (Biorad, Marne la Coquette, France) and further incubated for 48 h at 37 °C, or (iii) Columbia agar supplemented with 5% horse blood (Thermofisher Oxoid, Dardilly, France) and incubated for 48 h at 37 °C. P. aeruginosa CFUs were identified by MALDI-TOF mass spectrometry (Microflex LT; Bruker Daltonik GmbH, Bremen, Germany) according to the manufacturer’s recommendations. We stored each isolate of P. aeruginosa in brain-heart infusion broth supplemented with 30% glycerol at −80 °C until further analysis at the Centre de Ressources Biologiques - Filière Microbiologique de Besançon (Biobank BB-0033-00090).
Determination of susceptibility to antibiotics
We tested the activity of nine clinically relevant antibiotics (ceftazidime, cefepime, piperacillin-tazobactam, imipenem, meropenem, aztreonam, amikacin, tobramycin, and ciprofloxacin) against all P. aeruginosa isolates by the disk diffusion method on Muller-Hinton agar and interpreted it according to the recommendations of the European Committee on Antimicrobial Susceptibility Testing31.
Genome sequencing and phylogeny
Bacterial DNA was extracted using a QIAamp DNA Minikit (QIAgen, Hilden, Germany) according to the manufacturer’s recommendations. Isolates were first typed by MLST using PCR and sequencing according to the protocol of Curran et al., modified by ref. 32. We sequenced the genomes of all P. aeruginosa isolates using Illumina NextSeq high-output v2.5 technology (pair-end, 150 bp and coverage >240X; Microsynth, Balgach, Switzerland). Reads were subsampled to a coverage of 80X before genome assembly using Spades v3.13 with the optimized mode33. To build the phylogenetic tree of the species, we extracted the 5415 genomes of P. aeruginosa deposited at the NCBI by April 5, 2021. The sequence type (ST) of the strains was determined using pyMLST software34. We randomly selected one complete genome per ST to retain 753 genomes. Then, a core genome multilocus sequence typing (cgMLST) base with 3,867 genes was created. We built a phylogenetic tree from the multiple alignment of a subset of 3,786 genes present in >95% of the genomes using FastTree 2.1.10 with a GTR + G substitution model35.
To build the phylogenetic tree of ST308, we extracted the 95 genomes of P. aeruginosa ST308 deposited at the NCBI by April 5, 2021. We created a whole genome MLST (wgMLST) using genes of the strain Pa58 as a reference36. We selected 2937 genes present in 95% of the ST308 genomes and containing at least one variation for Bayesian phylogenetic analysis using, MrBayes v3.2.7a with a GTR + G + I substitution model37. We built a minimum spanning tree on ST308 strains described in our study from wgMLST allelic distances using Grapetree (v2.2)38.
Data availability
All raw reads generated were deposited under the BioProject accession number PRJNA807601 on the National Center for Biotechnology Information (NCBI) database. The data generated and/or analyzed in this study are available from the corresponding author upon reasonable request. All codes used in this study are available from the corresponding author on reasonable request or directly on the GitHub repository of the pyMLST software (github.com/bvalot/pyMLST)34.
References
Cuttelod, M. et al. Molecular epidemiology of Pseudomonas aeruginosa in intensive care units over a 10-year period (1998-2007). Clin. Microbiol. Infect. 17, 57–62 (2011).
Loveday, H. P. et al. Association between healthcare water systems and Pseudomonas aeruginosa infections: a rapid systematic review. J. Hosp. Infect. 86, 7–15 (2014).
Elborn, J. S. Cystic fibrosis. Lancet 388, 2519–2531 (2016).
Bedard, E., Prevost, M. & Deziel, E. Pseudomonas aeruginosa in premise plumbing of large buildings. MicrobiologyOpen 5, 937–956 (2016).
Jeanvoine, A. et al. Contamination of a hospital plumbing system by persister cells of a copper-tolerant high-risk clone of Pseudomonas aeruginosa. Water Res. 157, 579–586 (2019).
Abdouchakour, F. et al. Pseudomonas aeruginosa and Achromobacter sp. clonal selection leads to successive waves of contamination of water in dental care units. Appl. Environ. Microbiol. 81, 7509–7524 (2015).
Quick, J. et al. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: an observational study using whole-genome sequencing. BMJ Open 4, e006278 (2014).
Willmann, M. et al. Analysis of a long-term outbreak of XDR Pseudomonas aeruginosa: a molecular epidemiological study. J. Antimicrob. Chemother. 70, 1322–1330 (2015).
GBD 2019 Antimicrobial Resistance Collaborators. Global mortality associated with 33 bacterial pathogens in 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 400, 2221–2248 (2023).
Riquena, B. et al. Microbiological contamination of nebulizers used by cystic fibrosis patients: an underestimated problem. J. Bras. Pneumol. 45, e20170351 (2019).
HTM 04-01. In Health Technical Memorandum 04-01: Safe Water in Healthcare Premises (NHS England, 2016).
NF EN ISO 16266. Indice de classement: T 90-419: Qualité de l’eau - Détection et dénombrement de Pseudomonas aeruginosa - Méthode par filtration sur membrane (AFNOR Editions, 2008).
Wingender, J. & Flemming, H. C. Biofilms in drinking water and their role as reservoir for pathogens. Int. J. Hyg. Environ. Health 214, 417–423 (2011).
Bedard, E., Charron, D., Lalancette, C., Deziel, E. & Prevost, M. Recovery of Pseudomonas aeruginosa culturability following copper- and chlorine-induced stress. FEMS Microbiol. Lett. 356, 226–234 (2014).
Kim, J. S., Chowdhury, N., Yamasaki, R. & Wood, T. K. Viable but non-culturable and persistence describe the same bacterial stress state. Environ. Microbiol. 20, 2038–2048 (2018).
Li, L., Mendis, N., Trigui, H., Oliver, J. D. & Faucher, S. P. The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol. 5, 258 (2014).
Voigt, A. M. et al. The investigation of antibiotic residues, antibiotic resistance genes and antibiotic-resistant organisms in a drinking water reservoir system in Germany. Int. J. Hyg. Environ. Health 224, 113449 (2020).
Wang, L. et al. Assessment of the UV/chlorine process in the disinfection of Pseudomonas aeruginosa: efficiency and mechanism. Environ. Sci. Technol. 55, 9221–9230 (2021).
Hambsch, B., Hügler, M., Schönthal, M., Kempf, T. & Maier, M. Mikrobielle Belastung in Wasserzählern. Aqua Gas. 5, 22–28 (2016).
Martak, D. et al. High prevalence of Pseudomonas aeruginosa carriage in residents of French and German long-term care facilities. Clin. Microbiol. Infect. 28, 1353–1358 (2022).
Petitjean, M. et al. The rise and the fall of a Pseudomonas aeruginosa endemic lineage in a hospital. Microb. Genom. 7, 000629 (2021).
Michalska-Falkowska, A. et al. Emergence of Pseudomonas aeruginosa with class 1 integron carrying blaVIM-2 and blaVIM-4 in the University Clinical Hospital of Bialystok (northeastern Poland). Postepy Hig. Med. Dosw. 71, 589–594 (2017).
Gilarranz, R. et al. First detection in Europe of the metallo-beta-lactamase IMP-15 in clinical strains of Pseudomonas putida and Pseudomonas aeruginosa. Clin. Microbiol. Infect. 19, E424–E427 (2013).
Duchene, S. et al. Genome-scale rates of evolutionary change in bacteria. Microb. Genom. 2, e000094 (2016).
Dwidjosiswojo, Z. et al. Influence of copper ions on the viability and cytotoxicity of Pseudomonas aeruginosa under conditions relevant to drinking water environments. Int. J. Hyg. Environ. Health 214, 485–492 (2011).
Chew, K. L. et al. Challenge of drug resistance in Pseudomonas aeruginosa: clonal spread of NDM-1-positive ST308 within a tertiary hospital. J. Antimicrob. Chemother. 74, 2220–2224 (2019).
Perrin, Y., Bouchon, D., Hechard, Y. & Moulin, L. Spatio-temporal survey of opportunistic premise plumbing pathogens in the Paris drinking water distribution system. Int. J. Hyg. Environ. Health 222, 687–694 (2019).
Bourdin, T. et al. A high-throughput short sequence typing scheme for Serratia marcescens pure culture and environmental DNA. Appl. Environ. Microbiol. 87, e0139921 (2021).
NF EN ISO 19458. Qualité de l’eau - Échantillonnage pour analyse microbiologique. (AFNOR Editions, 2006).
Wang, H. et al. Methodological approaches for monitoring opportunistic pathogens in premise plumbing: a review. Water Res. 117, 68–86 (2017).
EUCAST. Breakpoint tables for interpretation of MICs and zone diameters. Version 10.0, www.eucast.org (2020).
van Mansfeld, R. et al. Pseudomonas aeruginosa genotype prevalence in Dutch cystic fibrosis patients and age dependency of colonization by various P. aeruginosa sequence types. J. Clin. Microbiol. 47, 4096–4101 (2009).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 70, e102 (2020).
Biguenet, A., Bordy, A., Atchon, A., Hocquet, D. & Valot, B. Introduction and benchmarking of pyMLST: an open-source software for assessing bacterial clonality using core genome MLST. Microb. Genom. 9, 001126 (2023).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490 (2010).
Espinosa-Camacho, L. F. et al. Complete genome sequences of four extensively drug-resistant Pseudomonas aeruginosa strains, isolated from adults with ventilator-associated pneumonia at a tertiary referral hospital in Mexico city. Genome Announc. 5, e00925–17 (2017).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Zhou, Z. et al. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res. 28, 1395–1404 (2018).
Rudra, B., Duncan, L., Shah, A. J., Shah, H. N. & Gupta, R. S. Phylogenomic and comparative genomic studies robustly demarcate two distinct clades of Pseudomonas aeruginosa strains: proposal to transfer the strains from an outlier clade to a novel species Pseudomonas paraeruginosa sp. nov. Int. J. Syst. Evol. Microbiol. 72, 005542 (2022).
Acknowledgements
This work was supported by the Commission des Examens de Laboratoire et des Innovations Analytiques (CELIA) of the Centre Hospitalier Universitaire of Besançon (X.B., D.H.).
Author information
Authors and Affiliations
Contributions
All authors contributed to the design or conduct of the study. A.J., A.A., and D.H. wrote the study protocol. D.H. and X.B. obtained funding. A.J., A.A., M.T., J.O., H.B., N.V.d.M.M., N.L., D.L., and M.E. collected the samples and epidemiological data. A.H., A.J., A.A., X.B., B.V., and D.H. performed or supervised the microbiological analyses. A.H., A.J., and B.V. provided a standardized procedure and performed genetic analyses for the whole-genome-sequencing. A.H., A.J., X.B., B.V., and D.H. drafted the manuscript and all authors reviewed and contributed to the manuscript. A.H. and A.J.: are considered “co-first author”.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Horikian, A., Jeanvoine, A., Amarache, A. et al. High-risk clones of Pseudomonas aeruginosa contaminate the drinking water networks of French cities. npj Clean Water 7, 35 (2024). https://doi.org/10.1038/s41545-024-00323-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-024-00323-8
