
Abstract
Antibody–drug conjugates (ADCs) are a promising cancer treatment modality that enables the selective delivery of highly cytotoxic payloads to tumours. However, realizing the full potential of this platform necessitates innovative molecular designs to tackle several clinical challenges such as drug resistance, tumour heterogeneity and treatment-related adverse effects. Several emerging ADC formats exist, including bispecific ADCs, conditionally active ADCs (also known as probody–drug conjugates), immune-stimulating ADCs, protein-degrader ADCs and dual-drug ADCs, and each offers unique capabilities for tackling these various challenges. For example, probody–drug conjugates can enhance tumour specificity, whereas bispecific ADCs and dual-drug ADCs can address resistance and heterogeneity with enhanced activity. The incorporation of immune-stimulating and protein-degrader ADCs, which have distinct mechanisms of action, into existing treatment strategies could enable multimodal cancer treatment. Despite the promising outlook, the importance of patient stratification and biomarker identification cannot be overstated for these emerging ADCs, as these factors are crucial to identify patients who are most likely to derive benefit. As we continue to deepen our understanding of tumour biology and refine ADC design, we will edge closer to developing truly effective and safe ADCs for patients with treatment-refractory cancers. In this Review, we highlight advances in each ADC component (the monoclonal antibody, payload, linker and conjugation chemistry) and provide more-detailed discussions on selected examples of emerging novel ADCs of each format, enabled by engineering of one or more of these components.
Key points
-
Antibody–drug conjugates (ADCs) are an effective cancer therapy, although responses to these agents are often limited by acquired resistance and treatment-related adverse effects.
-
Advances in the various ADC components (namely the antibody, linker, payload and conjugation chemistry) will be key to improving both the efficacy and safety of these agents.
-
To address these challenges, several novel ADC formats have been developed, including bispecific ADCs, probody–drug conjugates, immune-stimulating ADCs, protein-degrader ADCs and dual-drug ADCs.
-
Probody–drug conjugates are expected to have improved tumour specificity, whereas bispecific ADCs and dual-drug ADCs have the potential to combat drug resistance and tumour heterogeneity.
-
Integrating immune-stimulating ADCs and protein-degrader ADCs with current treatment regimens might enable multimodal treatment, potentially through several distinct mechanisms of action.
-
Patient stratification and biomarker identification will be crucial to maximize the clinical benefits of these emerging ADCs.
Similar content being viewed by others

Introduction
Antibody–drug conjugates (ADCs) have emerged as a promising class of cancer therapeutics. An ADC consists of a monoclonal antibody and a potent cytotoxic payload connected through a chemical linker. This molecular design combines the target specificity and long circulation half-life of an antibody with the high cytotoxic potency of antitumour agents that are too toxic for standalone use. Consequently, compared with conventional chemotherapies, ADCs can have enhanced antitumour efficacy, leading to improved clinical benefit and quality of life outcomes1,2,3,4. The considerable success of this emerging drug modality in patients with various types of cancer is demonstrated by the availability of 11 FDA-approved ADCs for at least 20 specific indications (Table 1). The level of interest in this modality has increased exponentially over the past few years, as demonstrated by the approval of four new ADCs within the past 3 years with more than 100 different ADCs currently being tested in clinical trials5. An economic analysis has revealed the rapidly growing size of the global market for ADCs: US $7.82 billion in 2022 with a projected compound annual growth rate of 11.2% from 2023 to 2030 (ref. 6).
Despite major progress in ADC development, the clinical potential of these drugs in patients with treatment-refractory cancers is often limited by various factors. Intratumour and intertumour heterogeneity is a major obstacle leading to poor therapeutic outcomes7,8. Tumour heterogeneity refers to the variability in genetic and phenotypic characteristics within a tumour (intratumour heterogeneity) or between tumours present within the same or different patients (intertumour heterogeneity), and both can cause variations in treatment response and lead to the emergence of ADC-resistant clones. Resistance to ADCs is intricately linked to the tumour heterogeneity; aggressive ADC therapy can create selective pressures that favour small subpopulations of resistant clones that harbour specific traits, including alterations in drug metabolism, mutations in the target proteins or their downstream signalling pathways, activation of alternative signalling pathways and/or the presence of cancer stem-like cells8,9. In addition to having activity across multiple tumour cell subpopulations, which minimizes the development of resistant clones, tolerability is a crucial parameter for the successful development of an ADC3. Indeed, many ADCs have been withdrawn either from clinical studies or from the market after initial approval owing to unacceptable toxicities and/or an overly narrow therapeutic window. An example includes the decision to withdraw the FDA-approved ADC gemtuzumab ozogamicin for CD33+ acute myeloid lymphoma (AML) from the market in 2010, and the subsequent re-approval of this agent at a lower dose in 2017 (ref. 10). Even successful ADCs that have been shown to provide clear clinical benefits to most patients can come with certain toxicity risks, as exemplified by interstitial lung disease and pneumonitis, which can be clinically serious and occasionally fatal, in patients receiving trastuzumab deruxtecan (T-DXd)3. Ingenious payload and linker designs, along with the identification of tumour-specific target antigens and effective biomarkers, will be crucial for the development of next-generation ADCs capable of overcoming these clinical challenges.
In this Review, we first provide a brief overview of the basic molecular design of an ADC and how each component (the antibody, linker, payload and conjugation chemistry) can affect the physicochemical and biophysical properties of the final product, including intracellular payload trafficking and metabolism, antitumour activity and safety profiles. Subsequently, we highlight and discuss selected examples of novel ADC designs that are currently in the early stages of preclinical and clinical development as next-generation cancer therapeutics, including bispecific ADCs, probody–drug conjugates (PDCs), immune-stimulating antibody conjugates (ISACs), protein degrader–antibody conjugates (DACs) and dual-payload ADCs. We aim to inform readers about the key design features necessary to generate effective and safe ADCs, as well as to provide an update on the extensive ongoing efforts to develop these agents and thus provide more and better treatment options for patients with cancer.
ADC design
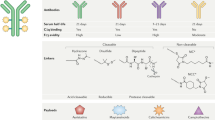
The principles of ADC design have evolved through the optimization of each structural component: namely the antibody, the cytotoxic payload and the chemical linker that connects these components (Fig. 1a). Extensive research efforts have provided insights into the implications of target selection and conjugation chemistries for the physicochemical properties as well as efficacy and safety of the ADC.

a, Schematic representation of an antibody–drug conjugate (ADC); each component (the antibody, payload, linker and conjugation chemistries) can all have important implications for the properties of the ADC. b, Chemical structures of non-cleavable and cleavable linkers. Although non-cleavable linkers remain attached to the payload structure upon intracellular release, cleavable linkers facilitate efficient release of the attached payloads in response to acidic pH, a reducing environment or degradation mediated by enzymes present within tumour cells or the tumour microenvironment (TME). c, Heterogeneous and homogeneous ADCs. Stochastic conjugation of payloads via lysine coupling or partial cysteine alkylation results in heterogeneous ADCs with variable drug to antibody ratios (DARs). By contrast, homogeneous ADCs with defined DARs are generated by full alkylation of interchain disulfides (as used in the manufacture of trastuzumab deruxtecan and sacituzumab govitecan) or site-specific conjugation through cysteine engineering techniques (such as THIOMAB), incorporation of reactive unnatural amino acids and following orthogonal coupling or enzymatic reactions. DAC, protein degrader–antibody conjugate; FcγR; Fcγ receptor; ISAC, immune-stimulating antibody conjugate; NK, natural killer; PDC, probody–drug conjugate; w/o, without.
Antibody and target selection
Among the many types of antibody available, humanized and fully human IgGs are most commonly used as the ADC backbone. Certain ADCs involve antibodies of the IgG4 subclass (such as gemtuzumab ozogamicin and inotuzumab ozogamicin)11; nonetheless, IgG1 antibodies are now preferentially used because of their general stability in the systemic circulation (reflecting an elimination half-life of 14–21 days) and robust engagement of innate immune cells, such as natural killer (NK) cells and macrophages, through interactions with Fcγ receptors (FcγRs)12. The use of human IgG1 also helps to reduce the overall immunogenicity of the ADC, which minimizes the risk of hypersensitivity reactions and the formation of anti-drug antibodies (ADAs)13. Most current ADCs maintain an N-linked glycan in N297 of the constant heavy chains to enable FcγR binding. However, interactions between such glycans and mannose receptors could drive nonspecific uptake of the ADC by hepatocytes14. Thus, using aglycosylated monoclonal antibodies might, in future, be a reasonable approach, depending on the specific pharmacokinetic or pharmacodynamic parameters of each ADC, such as payload potency, durability and biodistribution. Aglycosylation could be especially advantageous when the priority is to minimize the incidence of liver toxicities and inflammatory responses over enhancing potency. Aglycosylated antibodies can be vulnerable to structural distortion owing to thermal instability; nonetheless, data published in 2022 indicate that attaching small-molecule payloads to the CH2 domain of the Fc region can compensate for this instability15.
Antibodies that recognize antigens that are specifically expressed on cancer cell surfaces and are entirely absent from nonmalignant tissues are the ideal backbones for ADC construction, enabling the tumour-specific delivery of potent payloads. However, most ADC targets, including several that have already been successfully targeted (such as HER2 and TROP2), are also expressed by nonmalignant tissues to some extent16,17. Thus, even if ADCs are designed to target such validated molecules, both target-dependent and target-independent toxicities can still occur and might result in clinical holds or even the discontinuation of early phase testing (such as with MEDI4276 (ref. 18), XMT-1522 (refs. 19,20) and XMT-2056 (refs. 21,22) for patients with HER2+ breast cancer and PF-06664178 (ref. 23) for patients with TROP2-expressing solid tumours). To further enhance the tumour specificity of an ADC, antibodies capable of recognizing tumour-specific antigen variants with structural variations such as truncation, nicking (peptide bond cleavage caused by tumour-associated proteases) and other unique post-translational modifications have been explored (for example, EGFR variant III24,25, nicked TROP2 (refs. 26,27,28) and glycosylated PD-L1 (ref. 29)).
In addition to the antigen expression profile, ADC internalization and turnover rates can have important implications for efficacy30. Optimization of the binding affinity is also a crucial step towards maximizing ADC efficacy. Paradoxically, excessively strong antigen binding can lead to the retention of ADC molecules on the surfaces of tumour cells, thereby limiting the extent of tissue penetration (a phenomenon known as the binding-site barrier effect31,32). Thus, the antibody backbone of an ADC must be carefully selected, taking into account these various parameters to ensure optimal performance.
Payloads
The payloads used for ADCs are typically much more toxic than conventional chemotherapies, with sub-nanomolar or even picomolar levels of in vitro cytotoxicity observed as opposed to the micromolar levels of such activity seen with several common chemotherapies33,34. The payloads of current FDA-approved ADCs include anti-mitotic agents such as monomethyl auristatin E (MMAE), monomethyl auristatin F (MMAF) and the maytansine derivatives DM1 and DM4, DNA-damaging agents such as calicheamicins and pyrrolobenzodiazepine dimers (PBDs) and topoisomerase I inhibitors such as SN-38 and DXd. ADCs with other payloads including tubulysins (anti-mitotics)35,36,37,38,39, duocarmycins (DNA alkylators)40, PNU-159682 (a topoisomerase II inhibitor)41,42,43 and amanitin (an RNA polymerase II inhibitor)44,45,46 are currently being evaluated in preclinical and clinical studies. Beyond these cytotoxic payloads, immunomodulators47 and protein-degrader-recruiting molecules48 have emerged as promising novel payloads.
Most payload molecules have moderate to high levels of hydrophobicity, which is crucial in determining both the efficacy and the toxicity of the ADC. With several exceptions, such as MMAF, DM1 and amanitin, hydrophobic payloads can diffuse from target-expressing tumour cells into adjacent cells that might have limited or even no target expression, a phenomenon known as the bystander effect49,50. This effect is crucial for the successful eradication of heterogeneous tumours, in which both antigen-expressing and antigen-negative cells coexist. Despite this notable benefit, a hydrophobic payload can also have negative implications for ADC effectiveness. First, hydrophobic payloads can serve as good substrates for multidrug resistance proteins (such as MDR1, MRP1 and BCRP)51, thus diminishing the potency of certain ADCs against tumours that express these transporters. Second, hydrophobic ADCs tend to form aggregates, which can be rapidly cleared from the body52,53 and might be immunogenic54. Last, excessive hydrophobicity has been shown to facilitate liver uptake and cause hepatotoxicity55,56,57. ADC hydrophobicity is also a factor promoting nonspecific uptake through macropinocytosis, which might lead to ocular toxicities58 and thrombocytopenia59,60. As such, fine-tuning of the payload and ADC hydrophobicity are of paramount importance to overcome such issues while ensuring that the potential for bystander killing is retained. One approach to address this issue is to lower the number of conjugated payloads (the drug to antibody ratio (DAR)). However, lowering the DAR entails a reduction in antitumour activity, highlighting the importance of fine-tuning the DAR for each payload class to balance efficacy and toxicity. Installing hydrophilic masking groups such as long polyethylene glycol (PEG)53,61,62 or polysarcosine63,64,65,66 is another approach to this end, which enables the construction of high-DAR ADCs while avoiding some of the unwanted effects of high hydrophobicity.
Linkers
Chemical linkers have a pivotal role in enabling the cytotoxic payloads to remain attached to the antibody until the target is reached67,68 (Fig. 1b). Two primary linker types exist: non-cleavable and cleavable. Non-cleavable linkers are composed of stable bonds that resist proteolytic degradation, affording excellent stability in the systemic circulation. However, the release of the cytotoxic payloads bound by such non-cleavable linkers necessitates complete endocytosis and digestion of the antibody. This process is facilitated by cytosolic and lysosomal proteases, and results in the liberation of payload molecules that remain linked to a remnant amino acid residue from the degraded antibody (typically a cysteine or lysine). By contrast, cleavable linkers, which are preferentially used in current ADCs, are designed to be degraded by tumour-associated factors (such as the acidic and/or reducing conditions associated with most tumours or intracellular proteases). These linkers enable the efficient release of active payloads upon internalization into cancer cells, which results in cytotoxicity, thereby maximizing ADC potency and facilitating the bystander effect. Disulfide linkage and cathepsin-sensitive valine–citrulline dipeptides are commonly used for this purpose. However, cleavable linkers come with the risk of premature release of the payload into the circulation, which results in systemic toxicities and less-efficient payload delivery69. Therefore, careful linker design that strikes a balance between stability and efficacy is crucial. Research efforts from the past decade have focused on developing more-stable cleavable linkers, such as the GGFG tetrapeptide linker employed in T-DXd70, cathepsin-responsive tripeptide linkers71,72,73, as well as linkers cleaved by β-glucuronidase37,74,75, sulfatase76, phosphatase77 and legumain78,79.
Conjugation and homogeneity
In addition to the structural components discussed above, achieving high levels of homogeneity in bioconjugation is crucial to maximize the therapeutic window of an ADC. Most ADCs have traditionally been constructed using cysteine–maleimide alkylation or, less commonly, lysine–amide coupling (Fig. 1c). With some exceptions (such as T-DXd and sacituzumab govitecan), these stochastic conjugation methods result in a heterogeneous mixture of ADCs with variations in the payload attachment site and DAR. ADC heterogeneity often leads to less-efficient payload delivery owing to the rapid clearance of hydrophobic high-DAR components, necessitating strict production control to minimize such variations. To overcome these limitations, considerable efforts have been devoted to the development of site-specific conjugation methods for the production of homogeneous batches of ADCs with defined DARs. Notable examples of homogeneous conjugation technologies include full alkylation of interchain disulfides (used in T-DXd and sacituzumab govitecan), THIOMAB80 (a conjugation method that involves genetically incorporated cysteine residues), incorporation of non-naturally occurring reactive amino acids81,82,83, cysteine rebridging84,85,86,87,88,89, Fc-affinity tags90 and site-specific conjugation using various enzymes (such as engineered glycosidases91,92,93,94, transglutaminases95,96,97,98, formylglycine-generating enzymes99,100 and sortases101,102,103) (Fig. 1c).
Bispecific ADCs
As described previously, tumour heterogeneity and drug resistance often limit the antitumour activity of therapies directed towards a single target7,8. To address this challenge, bispecific antibodies have emerged as a method that enables simultaneous binding to two distinct target molecules and/or cells104,105. This approach has featured most prominently in bispecific T cell engagers, which elicit robust antitumour immune responses by tethering target-expressing tumour cells to T cells106,107. Bispecific ADCs that leverage this technology have received some attention as a potential avenue towards enhanced antitumour efficacy (Fig. 2). Of the various bispecific antibody formats developed104,108, human IgG1-based scaffolds are the most commonly used in the bispecific ADCs currently in development. The designs explored to date can be categorized into two types: bispecific ADCs that target different epitopes of the same antigen, which are also known as biparatopic ADCs; and bispecific ADCs that target two different antigens.

a, The biparatopic antibody–drug conjugates (ADCs) MEDI4276 and ZW49. MEDI4276 is an IgG1–single-chain variable fragment (scFv) fusion ADC that offers tetravalent binding to two distinct HER2 epitopes, extracellular domain 2 (ECD2) and ECD4, thereby promoting receptor clustering. This design enables increased binding affinity relative to trastuzumab, rapid internalization and enhanced potency. ZW49 is a heterodimeric ADC capable of bivalent binding to HER2 ECD2 and ECD4. b, Bispecific ADCs engineered to recognize EGFR in tandem with another antigen, potentially enhancing tumour specificity and enabling the eradication of a broader range of tumour cells. AZD9592 recognizes EGFR and MET, M1231 recognizes EGFR and MUC1 and BL-B01D1 recognizes EGFR and HER3.
Biparatopic ADCs
Data from previous studies indicate that the use of two anti-HER2 antibodies that bind to distinct epitopes can induce the formation of large receptor–antibody clusters on the cell surface, leading to endocytosis, lysosomal trafficking and downregulation of this target receptor109,110. Based on this observation, a multivalent biparatopic ADC that targets two different epitopes within HER2 hypothetically could have improved binding affinity, potentially resulting in more-efficient payload delivery, in particular to HER2low cancer cell populations111. To test this hypothesis, investigators generated a tetravalent HER2-targeting ADC named MEDI4276 by fusing a single-chain variable fragment (scFv) from trastuzumab with the N terminus of 39S, another anti-HER2 IgG1 antibody (Fig. 2a). A tubulysin derivative (AZ13599185), which is an anti-mitotic agent with low-picomolar levels of potency capable of bystander effects112, was then conjugated to this construct at a DAR of 4 using a stable linker via a site-specific conjugation method. As anticipated, this homogeneous biparatopic ADC had faster internalization kinetics and lysosomal trafficking via HER2 receptor clustering than was seen with either trastuzumab or the parent 39S antibody111. A later study revealed that the parent bispecific antibody targets HER2 extracellular domain 2 (ECD2) via the 39S Fab moiety and HER2 ECD4 by the trastuzumab scFv moiety113. Notably, MEDI4276 showed remarkable activity in mouse xenograft models of treatment-refractory HER2+ breast cancer, such as those featuring JIMT-1 cells, or the T-DM1-resistant NCI-N87 cell line, as well as in a panel of HER2low patient-derived xenograft (PDX) models characterized by intratumour heterogeneity111. Despite high levels of activity in preclinical models, MEDI4276 did not demonstrate a good efficacy–safety balance when tested clinically18. In patients with breast cancer, the overall response rate (ORR) was low (9.4%, 3 of 32 patients)18, which does not compare favourably with T-DXd (ORR 37% in patients with HER2low advanced-stage breast cancer in a separate study)114. The maximum tolerated dose (MTD) of MEDI4276 was determined to be 0.75 mg/kg every 3 weeks, although 7 of 12 patients who received this dose had one or more clinically serious and/or grade ≥3 adverse events, necessitating dose reduction18.
Another biparatopic HER2-targeting ADC, zanidatamab zovodotin (also known as ZW49), has been developed over the past few years115,116. ZW49 consists of a heterodimerized Fc region fused with an scFv that targets HER2 ECD4 and an Fab that targets ECD2 of the same protein, conjugated to an auristatin payload with an average DAR of 2 (ref. 117) (Fig. 2a). This asymmetrical structure enables bivalent HER2 binding, in contrast to the tetravalent binding accomplished by MEDI4276. Despite these differences in binding mode, ZW49 also induces receptor clustering and rapid internalization of HER2 (refs. 115,118). In a phase I dose-finding study testing ZW49, a recommended phase II dose (RP2D) of 2.5 mg/kg every 3 weeks was established119, which is comparable to the RP2Ds of other auristatin-based ADCs120,121,122. Among the 29 response-evaluable patients who received ZW49 under this dosing regimen, the confirmed ORR across multiple HER2+ advanced-stage cancer types was 28%, and the disease control rate was 72%. Only 9% of patients had grade ≥3 treatment-related adverse events (TRAEs), with clinically serious events in a further three patients. These results suggest that ZW49 has a manageable safety profile and promising antitumour activity in heavily pretreated patients. Both MEDI4276 and ZW49 are designed to recognize HER2 and promote receptor clustering, internalization and lysosomal trafficking. However, these biparatopic ADCs differ in terms of payload potency (tubulysin is more potent than auristatin in vitro), binding mode (tetravalent versus bivalent) and dissociation constant (137 pM111 versus 830 pM117). Although further investigations will be needed, these parameters might all be crucial in determining the therapeutic index of this novel class of ADCs.
REGN5093-M114, another biparatopic ADC, is designed to target two distinct epitopes of MET and is equipped with a maytansine derivative payload named M24 (ref. 123). REGN5093-M114 can be rapidly trafficked to recycling endosomes. However, in contrast to the biparatopic anti-HER2 ADCs described previously, the accumulation of this agent in late endosomes or lysosomes was not accelerated124,125. After multiple preclinical assessments, REGN5093-M114 is currently being tested in a phase I/II trial involving patients with advanced-stage non-small-cell lung cancer (NSCLC)126.
Bispecific ADCs targeting two different antigens
Simultaneous targeting of two different antigens using a bispecific ADC could offer multiple advantages. First, bispecific ADCs can recognize and kill a broader spectrum of tumour cells, including those from heterogeneous tumours. Second, if appropriate targets are chosen, bispecific ADCs might be capable of more tumour-specific binding owing to limited expression of both target antigens by nonmalignant cells and/or promoted payload uptake, thereby minimizing the risk of toxicities in nonmalignant tissues. Furthermore, engaging multiple antigens and/or cells simultaneously could elicit a synergistic effect that might not be feasible by targeting either antigen individually. Although these potential advantages have not yet been validated clinically, the use of bispecific ADCs could provide opportunities to improve efficacy and bypass the mechanisms of resistance that can arise during treatment with therapies directed towards a single target.
EGFR has been a focal point for the development of targeted therapies owing to the overexpression of this cell-surface receptor by various solid tumours. One promising approach that prolongs the clinical activity of EGFR-targeted therapy involves simultaneous inhibition of common resistance pathways, such as MET signalling127. An interim analysis of the phase III MARIPOSA study (NCT04487080) supports the potential of this approach; the combination of the EGFR–MET bispecific antibody amivantamab with the third-generation small-molecule EGFR inhibitor lazertinib resulted in a median progression-free survival (PFS) of 23.7 months, compared with 16.6 months with osimertinib, another third-generation inhibitor, as monotherapy128. Leveraging these findings, researchers have created bispecific ADCs that target EGFR and one other molecule that is preferentially co-expressed with EGFR on the tumour-cell surface129,130,131,132,133 (Fig. 2b). One group of investigators designed and evaluated a panel of EGFR–MET-targeted bispecific ADCs with varying binding affinities for EGFR129. Affinity-attenuated bispecific ADCs equipped with an MMAE payload showed a five- to sixfold greater therapeutic index than a cetuximab–MMAE ADC, based on differences in in vitro cytotoxic potency against EGFR and MET-expressing tumour cells and nonmalignant keratinocytes. Another line of research following the same concept led to the development of AZD9592, an EGFR–MET-targeted bispecific ADC equipped with a topoisomerase I inhibitor payload130,131. This agent demonstrated promising activity as monotherapy or in combination with osimertinib in PDX models of both EGFR-mutant and wild-type NSCLC, as well as head and neck squamous cell carcinoma (HNSCC). Importantly, AZD9592 was well tolerated in monkeys130. These promising preclinical results prompted the initiation of a phase I trial (NCT05647122).
Tumour-associated antigens other than MET have also been explored as combination targets for EGFR-targeting bispecific ADCs. For example, M1231 is a MUC1–EGFR-targeted bispecific ADC constructed from a heterodimeric antibody with an anti-MUC1 scFv and an anti-EGFR Fab domain132. The payload comprises SC209, a hemiasterlin derivative with anti-microtubule activity, conjugated through a cleavable valine–citrulline linker. Data from a preclinical study demonstrate superior internalization, lysosomal trafficking and improved therapeutic activity in PDX models of NSCLC and oesophageal squamous cell carcinoma (ESCC) compared with monospecific bivalent ADCs. A phase I dose-escalation study to evaluate the safety, pharmacokinetics and preliminary efficacy of M1231 (NCT04695847) was reportedly completed in June 2023, although detailed results do not appear to have been publicly disclosed thus far. Another example is BL-B01D1, a tetravalent bispecific ADC targeting EGFR and HER3 (ref. 133). This construct features a camptothecin derivative payload named ED04, which is attached by full cysteine conjugation and has a DAR of 8. Preclinical assessments confirmed the antitumour activity of this compound in mouse xenograft models of pancreatic or colorectal cancers compared with the corresponding ADCs targeting EGFR or HER3 only133. A phase I clinical trial has been initiated to test this agent in patients with unresectable locally advanced or metastatic solid tumours, including gastrointestinal and breast cancers (NCT05262491).
The bispecific ADC format has also been used to modulate intracellular processing and payload release kinetics. Internalized HER2 predominantly recycles back to the cell surface and is not efficiently trafficked to lysosomes134,135, thus hampering effective payload release following lysosomal degradation. Conversely, the prolactin receptor (PRLR) and amyloid precursor-like protein 2 (APLP2) have been shown to undergo rapid lysosomal trafficking upon endocytosis136,137,138,139. To capitalize on this feature, researchers have developed monovalent bispecific ADCs that target HER2 in combination with either PRLR140 or APLP2 (ref. 141). Both conjugates showed improved trafficking of ADC–HER2 complexes to lysosomes and comparable or slightly enhanced levels of in vitro potency relative to those of ADCs that target HER2 only. However, the HER2–APLP2-targeted ADC did not have improved activity in mouse xenograft models of breast cancer compared with its monospecific variant141. Multiple factors might contribute to the lack of improvement in antitumour activity, although the authors speculated that monovalent binding did not effectively induce HER2 dimerization, a key process for HER2 endocytosis, thus offsetting any potential benefits from accelerated lysosomal trafficking. This observation suggests that the efficacy of this construct might be improved by transforming the monovalent structure into a bivalent one.
In summary, bispecific ADCs offer promising therapeutic opportunities with the ability to target a wider spectrum of antigens, with improved activity, and possibly improved safety. However, this format also presents potential pitfalls that require careful consideration. One such example is the risk of unintended receptor activation and agonistic activity, as observed with certain anti-MET antibodies142,143,144 and EGFR–MET-directed bispecific antibodies145. Additionally, the expression ratios of the two target antigens can differ between tumours and patients, thus complicating the selection of potential responders. Careful evaluation of epitopes, binding modes and the underlying biology will therefore be crucial to achieve favourable treatment outcomes.
Probody–drug conjugates
Traditional ADCs that target receptors expressed not only on tumour cells but also on certain nonmalignant tissues (such as EGFR and TROP2) are often associated with unavoidable on-target off-tumour toxicities, leading to dose reductions or treatment discontinuation23,146,147,148. To address this issue, novel ADC designs that feature conditionally active antibodies (often referred to as probodies) have been developed (Fig. 3a,b). This design concept is inspired by prodrug formulations of small molecules, whereby pharmacologically inactive forms of the drugs are administered and then metabolized to their active forms in the circulation or in certain organs, leading to improved in vivo stability and/or specificity149,150,151. Probodies are IgG molecules that are either fused with self-masking moieties at the N terminus via cleavable spacers or designed with antigen-binding sites that undergo pH-dependent conformational changes, which reduces the target binding affinity of the IgG152,153,154,155,156,157,158,159. Upon reaching the tumour microenvironment (TME), either the masking moieties are removed or the antigen-binding sites change conformation in response to certain tumour-associated factors such as the abundance of proteases and acidic conditions, resulting in the localized restoration of the original target binding affinity of the antibody and payload release. This novel approach has the potential to enhance the therapeutic index of ADCs that might otherwise have excessive crossreactivity with nonmalignant tissues.

a, Schematic illustration of probody–drug conjugates (PDCs) incorporating coiled-coil peptide blocks (such as those that target CD19, integrin αVβ6 or CD3, which are all in preclinical development) or affinity binding peptides with protease-cleavable linkers (such as praluzatamab ravtansine, a CD166-targeted PDC, and CX-2029, a CD71-targeted PDC, both of which are being tested in early phase trials). Both types of masking moiety obstruct the epitope-binding regions within the Fab, thus inhibiting binding of the conjugate to its target receptor on nonmalignant tissues. Within the tumour microenvironment (TME), tumour-associated proteases degrade the spacer, thus activating the Fab moiety and facilitating specific tumour-cell targeting. b, PDCs that respond to variations in pH or concentrations of negatively charged ions. For pH-dependent PDCs, histidine residues within the deactivated Fab region become protonated in the mildly acidic TME but remain unprotonated under physiological conditions, prompting antigen binding via a conformational change. For PDCs that use a protein-associated chemical switch (PaCS), the Fab region and the antigen epitope are deactivated (blocked) by negatively charged ions such as chloride, bicarbonate and sulfide at the concentrations associated with physiological pH. These moieties become exposed in the TME owing to neutralization of some of these ions, thereby reactivating target binding.
PDCs with protease-sensitive self-masking moieties
In an early study153, investigators identified a cleavable peptide sequence (LSGRSDNH) sensitive to multiple proteases that are known to be minimally active in nonmalignant tissues but upregulated in the TME of various human cancers, including urokinase-type plasminogen activator160, membrane-type serine protease 1 (refs. 161,162) and legumain163. These investigators demonstrated that their anti-CD71 PDC, CX-2029, which contains another peptide spacer (ISSGLLSGRSDNP) sensitive to an undisclosed protease, and its ADC equivalent (both equipped with an MMAE payload at a DAR of ~2) had comparable levels of antitumour activity in mouse xenograft models of various solid tumours164. Based on the observed haematological toxicities, the preclinical MTDs of the PDC and the equivalent ADC in non-human primates were 6 and 0.6 mg/kg, respectively. These observations suggest that introduction of the masking moiety increased the therapeutic index by about tenfold.
Leveraging this innovation, these researchers have developed several first-in-class PDCs that are currently in preclinical development (CX-2051)165 and clinical development, including praluzatamab ravtansine ((formerly known as CX-2009) NCT03149549 and NCT04596150) and CX-2029 (NCT03543813)164,166,167,168 (Fig. 3a). Praluzatamab ravtansine is an anti-CD166 PDC equipped with the microtubule inhibitor DM4 (refs. 166,168). CD166 is broadly expressed in many nonmalignant tissues169,170, thereby providing an ideal target for the PDC approach. In a phase I study, 99 patients with metastatic solid tumours, including breast cancer (46%), epithelial ovarian carcinoma (22%) and NSCLC (13%), received praluzatamab ravtansine168. Tumour regression was observed at doses of ≥4 mg/kg. Among patients with HR+, HER2-nonamplified breast cancer, two partial responses (in 9% of the cohort) were observed, with stable disease in a further ten patients (45%). Unconfirmed partial responses were reported in three patients with triple-negative breast cancer (30%). Dose-limiting toxicities such as keratitis (in 9% of patients), increased serum aspartate aminotransferase (8%) and alanine aminotransferase (5%) levels, and anaemia (5%) were observed with doses at 8 mg/kg every 3 weeks and 6 mg/kg every 2 weeks, therefore a RP2D of 7 mg/kg every 3 weeks was selected. Despite this recommendation, the ongoing phase II trial to test this PDC (NCT04596150) is being conducted with doses of 6 mg/kg every 3 weeks. CX-2029, an anti-CD71 PDC with an MMAE payload is currently being tested in a phase I trial involving patients with various advanced-stage solid tumours such as NSCLC (20%), HNSCC (18%) and colorectal cancer (16%)167. Among 45 patients who received CX-2029, relevant antitumour activity was observed in 16 patients (3 partial responses and stable disease in 13 patients); 3 mg/kg every 3 weeks was selected as the RP2D. The clinical utility of this PDC design will be fully elucidated by the outcomes of patients enrolled in the ongoing clinical trials.
A major challenge associated with the development of PDCs is the need for extensive screening and optimization of the self-masking peptide sequences for each antibody–target epitope combination. As a more generalizable approach, a novel PDC format using a human leucine zipper heterodimeric coiled-coil domain as a stearic masking group has been developed171 (Fig. 3a). This rigid peptide structure is fused to the N terminus of both the light and heavy chains of the antibody via matrix metalloproteinase (MMP)-cleavable peptide sequences, thus sterically inhibiting antibody–target binding. These researchers demonstrated the universal applicability of this technology for tumour-specific activation; hBu12 (anti-CD19), rituximab (anti-CD20), trastuzumab (anti-HER2), h15H3 (anti-integrin αVβ6) and 145-2C11 (anti-mouse CD3) antibodies engineered with the same blocking unit showed minimal in vitro binding to their target antigens (at concentrations of up to 2 μM) but regained 80- to 1,000-fold greater binding affinity upon the introduction of MMPs and subsequent cleavage of the masking domain171. In the same study, conditionally masked hBU12, h15H3 and 145-2C11 PDCs demonstrated improved circulation half-lives and greater antitumour activity in xenograft models compared with their unmasked equivalents.
PDCs with pH-responsive antigen-binding sites
The TME is usually slightly more acidic (pH 6.0–6.8) than most nonmalignant tissues (pH 7.3–7.4). This difference in pH can be used to enable conditional activation of ADCs owing to a reversible conformational change in the antigen-binding site (Fig. 3b). Incorporating weakly basic histidine residues into the binding regions of the antibody is a common method of conferring such pH-dependent activation. Of the various pH-dependent ADCs developed to date (including those that target EGFR172,173, HER2 (ref. 174), AXL175 and ROR2 (ref. 176)), the MMAE-based EGFR-targeted PDC HTI-1511 is the most notable example with positive preclinical data172,173. The parent antibody of HTI-1511 has a more than tenfold greater binding affinity for EGFR at pH 6.0–6.5 than at pH 7.4. This antibody did not show detectable binding in mouse xenograft models injected with EGFR-expressing cells derived from nonmalignant human foreskins but maintained levels of binding affinity for EGFR-expressing A431 xenografts comparable to that of cetuximab172. HTI-1511 also induced substantial inhibition of, and even regression of, tumour growth in cetuximab-resistant PDX models and in those harbouring KRAS or BRAF mutations172. HTI-1511 showed favourable tolerability at doses of up to 8 mg/kg in cynomolgus monkeys, indicating a promising clinical safety profile172. Despite these promising preclinical data, the clinical development of HTI-1511 has seemingly not progressed since 2018. This lack of progress could be attributed to a range of factors, including potential competition in the EGFR ADC space, unforeseen technical challenges in the production and scale-up process or a strategic pivot in the company’s development priorities in light of emerging data or market considerations177.
As another example of a PDC capable of responding to the TME, investigators developed pH-dependent PDCs based on a mechanism termed protein-associated chemical switch (PaCS)178. The complementarity-determining regions within PaCS-based PDCs are engineered to enable interactions with abundant ions, including sodium chloride, bicarbonate and hydrogen sulfide. At a pH of ~7.4, negatively charged forms of these molecules are present at concentrations sufficient to suppress antigen binding via interactions with the positively charged complementarity-determining regions. However, concentrations of these ions are lower in the more acidic TME, enabling ion concentration-dependent activation of target binding. The requirement for both low pH and specific ion concentrations potentially offers higher tumour specificity than that provided by pH-dependent activation alone. Various PDCs have been developed using this platform and are being tested in phase I/II trials involving patients with various advanced-stage solid tumours, including the AXL-targeted PDC BA3011 (NCT03425279, NCT04681131) and the ROR2-targeted agent BA3021 (NCT03504488, NCT05271604)179. Other PDCs involving PaCS technology, including BA3361 (targeting nectin-4) and BA3151 (targeting B7-H4), are in preclinical development179.
In summary, PDC platforms hold great promise in enabling the targeting of antigens that are otherwise inaccessible owing to undesirable levels of expression in nonmalignant tissues. However, the identity of specific cancer subtypes that can be effectively targeted using PDCs featuring certain conditional activation mechanisms remains to be fully clarified. In-depth preclinical and clinical studies will be necessary to determine the optimal use of such novel ADCs and to develop evidence-based guidelines for the identification of patients who will benefit most from this approach.
ISACs
The transformative advances in cancer immunotherapy from the past decade, exemplified by the success of immune-checkpoint inhibitors targeting the PD-1 and/or CTLA4 signalling pathways, have sparked renewed interest in this field180. In the context of innate immune activation triggered by the release of damage-associated molecular patterns (DAMPs) from tumour cells, immunological adjuvant molecules that interact with pattern-recognition receptors (PRRs) have emerged as a focal point in cancer drug development181. Within this framework, researchers have developed various agonists for Toll-like receptors (TLRs)182,183 and, more recently, stimulator of interferon genes (STING)184. When activated, these pivotal mediators can initiate signalling cascades that lead to cytokine production, which can then orchestrate and/or amplify an antitumour immune response. Despite these agents being attractive alternatives to conventional chemotherapy, systemic administration can cause severe adverse events owing to hyperactivation of the immune system185,186. Consequently, drugs of this class are typically administered as intratumoural injections to minimize the risk of systemic toxicities187. Currently, neither TLR nor STING agonists have gained clinical approval and their efficacy in clinical trials has been variable, underscoring the challenges in harnessing innate immunity for cancer treatment.
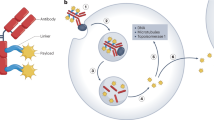
Among various attempts to deliver PRR agonists specifically to tumours188,189, systemic administration of antibodies conjugated with specific PRR agonists has emerged as a promising method of localized activation of innate immunity (Fig. 4a). Preclinical evaluation has demonstrated the potential advantages of ISACs over conventional ADCs that carry cytotoxic payloads47: first, the ISAC-mediated antitumour response can develop against multiple tumour-associated DAMPs; second, ISAC-mediated immune stimulation eventually activates not only antigen-presenting cells (APCs) but potentially also other tumour-infiltrating immune cells such as T cells; and third, ISACs elicit an immunological memory effect throughout the cellular immune response, providing durable antitumour effects and reducing the risk of recurrence. These features have yet to be validated clinically; nonetheless, the multimodal antitumour immunity provided by ISACs suggests the potential to reduce the risk of, and/or delay the onset of, acquired resistance.

General mechanism of action of immune-stimulating antibody conjugates (ISACs). ISACs bound to a tumour-specific antigen are internalized into antigen-presenting cells (APCs) through interactions with Fcγ receptors. This internalization leads to the intracellular release of tumour-associated peptides and conjugated pattern-recognition receptor (PRR) agonists, such as Toll-like receptors (TLRs) and stimulator of interferon genes (STING). Release of these mediators synergistically triggers the release of pro-inflammatory cytokines, attracts immune cells to the tumour site and enhances antigen presentation, resulting in a robust, specific antitumour immune response. If the conjugated PRR agonists are sufficiently hydrophobic and can engage the bystander effect, ISACs can also deliver these agonists to APCs through internalization into tumour cells and following intercellular diffusion (panel a). Schematic representations of BDC-1001, a HER2-targeted ISAC with a TLR7/8 agonist payload (panel b); TAC-001, a CD22-targeted ISAC with a chemically modified CpG TLR9 agonist payload (panel c); and XMT-2056, a HER2-targeted ISAC with a dimeric amidobenzimidazole (diABZI) STING agonist payload (panel d).
ISACs with TLR7/TLR8/TLR9 agonist payloads
Among all the TLRs characterized to date, TLR7, TLR8 and TLR9 are the major targets of most ISACs developed thus far. These TLRs are intracellular PRRs found in the endosomes of APCs, such as dendritic cells and macrophages. Activation of these endosomal TLRs in the presence of cancer promotes the presentation of tumour-associated DAMPs by APCs, resulting in robust antitumour effects via activation of both the innate and adaptive immune responses181.
In 2015, investigators described the activity of an anti-CD20 ISAC developed by conjugating TLR7 agonists to rituximab190. In vitro testing showed that conjugation did not impair the antigen-binding capacity or specificity of rituximab, or the TLR-stimulating activity of the agonists. This early research paved the way for the development of various TLR7/8-activating ISACs. In a subsequent seminal study, researchers described a HER2-targeted ISAC with promising in vivo activity47. Either T785 (a TLR7–TLR8 dual agonist) or CL264 (a TLR7-specific agonist) were conjugated to the anti-HER2 antibody trastuzumab using a non-cleavable linker, yielding HER2-targeted ISACs with an average DAR of approximately 2. Either a single dose or multiple doses of these ISACs at 5 mg/kg had moderate to high antitumour activity along with robust myeloid activation and cytokine release in mouse xenograft models of HER2+ breast cancer. Notably, ISACs constructed using an anti-rat HER2 antibody almost completely suppressed tumour growth in syngeneic mouse models when administered at 5–10 mg/kg every 5 days. Furthermore, mice exposed to this ISAC were protected from tumour regrowth following rechallenge with a HER2− form of the parental tumour cell line. This result indicates the presence of robust immunological memory not only against HER2 but also against other DAMPs.
After these encouraging preclinical results, the anti-HER2 TLR8 ISAC SBT6050 was commercialized as pertuzumab zuvotolimod, consisting of a TLR8 agonist conjugated to pertuzumab via a cleavable linker. This agent was tested as a monotherapy and in combination with an anti-PD-1 antibody pembrolizumab or cemiplimab in a phase I study (NCT04460456) and with other HER2-targeted therapies in a phase I/II study (NCT05091528). Despite much anticipation, cytokine-related adverse events curtailed the range of doses that could be administered with pembrolizumab191. The lack of any noteworthy single-agent activity further confounded this challenge, ultimately leading to the discontinuation of these studies192. Another HER2-targeted ISAC, NJH395, comprising a TLR7 agonist conjugated to an anti-HER2 antibody via a non-cleavable linker was evaluated in a phase I trial (NCT03696771)193. However, this study was also discontinued after completion of the single-ascending dose part owing to insufficient efficacy and the prevalence of TRAEs, including cytokine-release syndrome (in 10 of 18 patients) and ADA formation in all patients194. Other examples of HER2-targeted ISACs include BDC-1001, a trastuzumab-based ISAC equipped with a TLR7/8 agonist via a non-cleavable linker195 (Fig. 4b). BDC-1001 is currently being tested in a phase I/II trial involving patients with advanced-stage HER2-expressing solid tumours, either as a monotherapy or in combination with the anti-PD-1 antibody nivolumab196. A phase II trial for BDC-1001 in combination with pertuzumab is also ongoing (NCT05954143). Unlike SBT6050 and NJH395, BDC-1001 has demonstrated promising preliminary results: these revealed some evidence of clinical activity (four confirmed durable partial responses and stable disease ≥6 months in a further ten patients), with minimal clinically serious toxicities and no ADA formation across all tested dose levels (0.15–20 mg/kg)197. In addition to HER2-targeted ISACs, ISACs targeting CEACAM5 (ref. 198) and PD-L1 (ref. 199) have shown encouraging antitumour activity in preclinical studies.
Data from subsequent studies have underscored the promise of TLR7/8-activating ISACs. An anti-PD-L1 ISAC was generated by conjugating D18, a bifunctional immunoregulatory agent, to an anti-PD-L1 antibody via a redox-cleavable linker using THIOMAB technology at a DAR of 2 (ref. 200). This ISAC induced a robust antitumour immune response through TLR7/8 activation with substantial suppression of tumour growth in mouse syngeneic models. Elsewhere, investigators developed anti-HER2 ISACs conjugated with various imiquimod-based TLR7 agonists with a DAR of 8 (ref. 201). Three doses of this ISAC induced tumour regression in mouse xenografts bearing HER2+ HCC1954 tumours (three of five mice at 3 mg/kg and all mice at 10 mg/kg).Unlike other ISACs, these conjugates were non-glycosylated (Fcγ null) constructs that are unable to directly engage innate immune cells. Nonetheless, the payloads were released from tumour cells and were able to potently activate tumour-associated macrophages in vitro via a bystander effect. Considering that Fcγ-mediated uptake of ADCs by APCs could lead to the development of ADAs, this unique conjugate design could facilitate the creation of safer ISACs.
TAC-001 is an anti-CD22 antibody equipped with a synthetic CpG oligonucleotide payload, which is a potent TLR9 agonist, at a DAR of ~1 (refs. 202,203) (Fig. 4c). When administered intravenously, TAC-001 delivers the payload to CD22-expressing B cells, which initiates TLR9 signal transduction, B cell activation and a cascade of immune responses. This process also activates other TLR9-expressing immune cells, including dendritic cells and monocytes. Data from preclinical studies demonstrate that two 10 mg/kg doses administered 3 days apart achieve substantial suppression of tumour growth and strong memory responses in multiple syngeneic tumour models, indicating that TAC-001 promotes robust innate and adaptive antitumour immune responses202. This agent is currently being tested in a phase II trial involving patients with various advanced-stage solid tumours (NCT05399654)204.
ISACs with STING agonist payloads
The cGAS–cGAMP–STING axis is activated by exposure to foreign DNA derived from microbial pathogens and/or dying tumour cells, leading to the production of type I interferons and activation of innate immunity205. Data from several studies indicate that STING signalling is crucial to induction of a robust T cell-mediated antitumour immune response206,207 along with T cell infiltration into the TME208,209. EGFR-targeted ISACs have been produced by conjugating IMSA172, a cGAMP analogue, to an anti-EGFR antibody via a cleavable valine–citrulline linker at a DAR of 3–4 (ref. 210). Systemic administration of these ISACs was well tolerated and resulted in potent antitumour activity in EGFR-expressing mouse xenograft models with three doses of 200 μg (approximately 8–10 mg/kg) administered at 3-day intervals. The antitumour activity of these ISACs was further enhanced by combination with an anti-PD-L1 antibody. These investigators also confirmed robust activation of T cells, NK cells, dendritic cells and NKT cells, and macrophage polarization.
A potent ISAC, named XMT-2056, armed with a non-cGAMP-type STING agonist has been developed over the past few years21,211,212. This construct leverages a derivatized version of the novel, highly potent dimeric amidobenzimidazole STING agonist diABZI213 conjugated to an anti-HER2 antibody using a hydrophilic linker at a DAR of 8 (Fig. 4d). Remarkably, a single 1 mg/kg dose of this ADC and an anti-rat HER2 surrogate induced durable and complete tumour regression in HER2+ mouse xenografts as well as in a mouse syngeneic model established using a rat HER2-expressing subline21. When co-administered with either trastuzumab, pertuzumab, T-DXd or an anti-PD-1 antibody, XMT-2056 showed further enhanced efficacy even at lower dosing levels21. This ISAC was well tolerated in non-human primates at 9 mg/kg212. XMT-2056 is currently being tested in a phase I study involving patients with advanced-stage or recurrent HER2-expressing solid tumours (NCT05514717). Unfortunately, a patient injected with XMT-2056 at the initial dosing level in a dose-escalation study had a fatal (grade 5) drug-related adverse event, leading to a clinical hold in March 2023 (ref. 22). The FDA lifted this hold in October 2023 after the investigators lowered the starting dose214.
In conclusion, ISACs are a promising new class of cancer immunotherapy. Some of the initial clinical trial results are encouraging, and further studies are currently exploring their potential in various cancer types, both as monotherapies and in combination with other modalities. However, continued research will be crucial to fully optimize both the efficacy and the safety profiles of these agents. Specifically, judging from the apparently lower efficacy reported for TLR-activating ISACs compared with traditional ADCs, further improvements in potency might be necessary. Conversely, the discontinuation of the phase I trial testing NJH395 (ref. 194) underscores the crucial importance of improvements that reduce the risks of ADAs and excessive inflammatory responses. Although further investigation is needed, the clinical hold previously placed on the study testing the highly potent agent XMT-2056 (ref. 22) suggests that the toxicity of STING-activating ISACs might not be accurately estimated based on data from preclinical models, probably owing to unknown patient-specific factors. Overall, a better understanding of the pharmacological effects of ISACs on the human immune system along with appropriate patient stratification will be crucial for the development of this class of ADCs.
Antibody-based protein degraders
DACs, which carry a proteolysis-targeting chimera (PROTAC) instead of the traditional cytotoxic payload, are a novel class of targeted therapies that capitalize on the breakthrough technology behind PROTACs48,215 (Fig. 5a). PROTACs are heterobifunctional molecules that consist of two ligands interconnected via a linker. One ligand targets a protein of interest (POI) while the other ligand engages an E3 ubiquitin ligase, such as the von Hippel–Lindau protein (VHL) or cereblon (CRBN)216. This ingenious molecular design enables PROTACs to simultaneously bind to both the POI and an E3 ligase, triggering targeted ubiquitylation followed by degradation by the proteasome. In contrast to classical inhibitors, ligand binding to the POI does not require antagonistic activity. Consequently, this unique approach enables the modulation of various proteins that were previously deemed ‘undruggable’ owing to the challenges in identifying small molecules with both inhibitory activity and a sufficiently high binding affinity (more-detailed descriptions of the development of PROTACs and clinical progress with these agents are available elsewhere217,218,219). The DAC format has the potential to further advance the clinical utility of this novel modality, offering high levels of tumour specificity and durable activity by harnessing the power of antibody-based drug delivery.

General mechanism of action of protein degrader–antibody conjugates (DACs). The monoclonal antibody moiety recognizes a tumour-associated antigen, triggering internalization of the DAC–antigen complex. The linker degrades under proteolytic, acidic and/or reducing conditions, thus releasing the conjugated proteolysis-targeting chimera (PROTAC) molecules into the cytoplasm. This leads to ubiquitylation of a protein of interest (POI) via engagement of the E3 ligase, resulting in degradation of the POI (panel a). Schematic illustration of a GNE-987-containing DAC that targets BRD4, which is being developed for patients with CLL1+ acute myeloid leukaemia (AML) (panel b), and ORM-5029, which targets the G1 to S phase transition 1 protein (GSPT1) and is currently being tested in patients with HER2+ breast cancer (NCT05511844) (panel c). The E3 ligand (von Hippel–Lindau protein (VHL) or cereblon (CRBN)) and POI-targeting ligand moieties are highlighted in blue and yellow, respectively.
Bromodomain and extra-terminal motif (BET) proteins, particularly bromodomain-containing protein 4 (BRD4), have a crucial role in the epigenetic regulation of acetylated histones across various tumour types. Although many BET inhibitors have been developed, the clinical success of these agents has been hampered largely by insufficient potency and dose-limiting toxicities220,221. Consequently, these proteins have emerged as attractive targets for PROTAC-based cancer therapies222. A report published in 2020 describes the development of a potent chimeric BET degrader named GNE-987 (ref. 223). This compound consists of a ligand for BRD4 and a VHL-recruiting molecule. GNE-987 demonstrated potent degradation of BRD4 in EOL-1 cells (half-maximal degradation concentration (DC50) of 0.03 nM) and excellent in vitro potency in two cell lines (half-maximal inhibitory concentration (IC50) of 0.02 nM in EOL-1 and 0.03 nM in HL-60); however, it was ineffective in in vivo models owing to the unfavourable drug metabolism and pharmacokinetic profile223. To address this issue, researchers converted GNE-987 into a homogeneous DAC by linking the hydroxyl group of GNE-987 to six cysteines genetically incorporated into an anti-CLL1 antibody using labile carbonate linkage223 (Fig. 5b). The hydroxyl group is crucial for binding to VHL224,225, and this conjugate design allows for the intracellular activation of the conjugated GNE-987 once it is delivered to tumour cells and the carbonate linkage is hydrolysed. As expected, a single intravenous administration of the DAC at 10 mg/kg enabled persistent in vivo exposure and remarkable suppression of tumour growth in subcutaneously xenografted HL-60 and EOL-1 mouse models of AML223. Although only in vitro data have been presented, a HER2-directed, BRD4-degrading DAC has also been developed based on a similar molecular design226. Both of these conjugates contain either labile carbonate or ester linkages that enable payload release upon intracellular processing, which can lead to premature linker degradation and undesired payload release into the circulation as observed for sacituzumab govitecan, a carbonate-linked ADC227,228,229. To mitigate this risk, carbamate linkage is often used to ensure stability. For example, investigators synthesized and evaluated a panel of BRD4–VHL PROTAC derivatives containing an amine, enabling attachment to a carbamate-based self-immolative linker230,231. These DACs had comparable levels of in vitro cytotoxicity and in vivo antitumour activity in the subcutaneously xenografted HL-60 mouse model. Although no direct comparisons with carbonate linkers were made, an aniline-linked carbamate DAC demonstrated no change in average DAR 8 days after intravenous injection into mice. Elsewhere, a HER2-targeted DAC equipped with a BRD4–CRBN PROTAC has been developed using a similar molecular design232.
Numerous intracellular proteins beyond BRD4 have been extensively targeted preclinically using DAC-based therapies, such as oestrogen receptor-α (ERα)233, TGFβ receptor 2 (ref. 234) and the chromatin regulatory protein SMARCA2 (also known as BRM)235. Two DACs designed to degrade the G1 to S phase transition 1 (GSPT1) protein are showing early signs of clinical potential236,237,238,239. These DACs use a highly potent CC-885-derived GSPT1–CRBN degrader as the payload. This degrader acts a ‘molecular glue’ that modifies the protein surface to enable new protein–protein interactions between GSPT1 and CRBN. This interaction creates substantial disruption of protein synthesis, leading to apoptosis240,241,242. These investigators conjugated the CC-885 derivative to the anti-HER2 antibody pertuzumab using a valine–citrulline linker, resulting in a DAR of 4 (ref. 243) (Fig. 5c). This HER2-directed DAC, named ORM-5029, had 100- to 1,000-fold greater in vitro cytotoxicity than could be achieved using other GSPT1 degraders and traditional ADCs, including T-DM1 and T-DXd236,237. A single 10 mg/kg dose of ORM-5029 provided excellent inhibition of tumour growth in the MDA-MB-453 mouse xenograft model, which was comparable to that of an equivalent dose of T-DXd. A phase I trial testing ORM-5029 in patients with advanced-stage HER2+ solid tumours is ongoing (NCT05511844). Outcomes of this study will be crucial in gauging the clinical potential of this novel ADC class. ORM-6151, a CD33-targeted DAC for patients with AML, has been developed by the same company. This DAC has demonstrated antigen-dependent in vitro cytotoxicity comparable to that of the FDA-approved anti-CD33 ADC gemtuzumab ozogamicin with superior activity, including complete eradication of all tumour cells in nine of nine animals with a single dose at 3 mg/kg in an MV4-11 subcutaneous mouse xenograft model of AML238,239. Notably, even a 0.1 mg/kg single dose demonstrated effective disease control in a disseminated version of the MV4-11 xenograft model. Toxicity assessments revealed limited ORM-6151-induced damage to nonmalignant bone marrow progenitor cells, whereas gemtuzumab ozogamicin resulted in substantial toxicity. These results highlight the clinical potential of ORM-6151.
In addition to DACs, which are designed to degrade specific cytosolic proteins as described above, several groups have developed antibody-directed cell-surface protein degraders, including antibody-based PROTACs (AbTACs)244, proteolysis-targeting antibodies (PROTABs)245 and lysosome-targeting chimeras (LYTACs)246,247. AbTACs and PROTABs are heterodimeric bispecific antibodies that consist of a cell-surface POI recognition region (recognizing targets such as PD-L1 (refs. 244,245), IGF1R245 and HER2 (ref. 245)) and a region that recognizes transmembrane E3 ubiquitin ligase proteins, such as RNF43 or ZNRF3 (ref. 248). LYTACs consist of an antibody that recognizes a cell-surface POI conjugated to a ligand capable of promoting lysosomal degradation of the antibody–target protein complex through interactions with lysosome-associated membrane proteins, such as oligomeric mannose 6-phosphate246 or trimeric N-acetyl galactose247. All of these novel entities have demonstrated remarkable preclinical activity in targeted protein knockdown experiments.
In summary, DACs and other antibody-directed protein degraders are pioneering technologies with the potential to offer unique therapeutic interventions for patients with cancer. Nonetheless, most of these entities are still in the early preclinical stages of development, and further medicinal chemistry studies and other preclinical evaluations are required to identify safe and potentially effective conjugates that can advance to clinical testing. Specifically, the payloads used for DACs are generally hydrophobic, leading to an excessively hydrophobic DAC. This hydrophobicity might be exacerbated by the need for relatively high DARs (6 or higher) to ensure sufficient cytotoxicity, owing to the generally lower potency of protein degraders relative to traditional ADC payloads. These issues can compound the negative effects of hydrophobicity on the pharmacokinetics and toxicity profiles of these agents. Thus, novel linker designs and modifications of the degrader molecules that address these issues could substantially advance this already promising modality.
Dual-drug ADCs
Most solid tumours are comprised of heterogeneous cancer cell subpopulations with varying gene expression profiles and levels of sensitivity to drugs with various mechanisms of action7,249. As such, relying on a single agent for tumour eradication can exert selective pressures that enable the survival and growth of insensitive tumour cell subpopulations, leading to disease relapse and acquired resistance. Thus, combination regimens that involve multiple agents with differing modes of action are commonly adopted in clinical practice. Advances in conjugation chemistry (such as orthogonal protection of conjugation handles250 and dual click reactions72) have demonstrated the feasibility of delivering combinations of cytotoxic agents using ADCs equipped with two distinct payloads, often called dual-drug ADCs (Fig. 6a). Dual-drug ADCs have the potential to elicit additive or synergistic effects as a single agent and overcome resistance in patients with treatment-refractory tumours while maintaining a simple dosing regimen72,250. Although potentially advantageous in overcoming tumour heterogeneity and resistance, it should be noted that dual-drug ADCs might also cause additive or synergistic toxicities. Several orthogonal conjugation strategies have been developed to generate dual-drug ADCs with high levels of homogeneity and defined DARs.

a, Structure and conjugation properties of current dual-drug antibody–drug conjugates (ADCs). These include ADCs with two cytotoxic payloads (such as monomethyl auristatin E (MMAE) plus monomethyl auristatin F (MMAF) or MMAE plus SG3457), or ADCs that combine a cytotoxic and an immunomodulatory payload (such as hemiasterlin plus a Toll-like receptor (TLR) agonist). Dual-drug ADCs are generated either using bifunctional branched linkers or by the combination of orthogonal conjugation of conventional linear linkers to two different amino acid residues. b, Synthesis of a CD30-targeted dual-drug ADC developed by Levengood et al.250. These investigators used a branched linker with two cysteines protected by two orthogonal groups (SiPr and Acm). Conjugation was achieved using stepwise orthogonal deprotection and payload installation resulting in a homogeneous dual-drug ADC equipped with MMAE and MMAF at a drug to antibody ratio (DAR) of 8 + 8. c, Synthesis of HER2-targeted dual-drug ADCs developed by Yamazaki et al.72. These investigators developed various branched linkers that contained orthogonal click reaction handles (azides and tetrazines (Tet)). Enzymatic linker conjugation followed by sequential click reactions with MMAE and MMAF modules yielded a panel of homogeneous dual-drug ADCs with defined DARs (MMAE–MMAF 2 + 2, 4 + 2 and 2 + 4). mAb, monoclonal antibody.
An efficient method of producing dual-drug ADCs using branched chemical linkers containing two orthogonally masked cysteine residues was reported in 2017 (ref. 250) (Fig. 6b). By sequentially coupling the payloads and unmasking the cysteine residues, these investigators were able to homogeneously conjugate MMAE and MMAF to an anti-CD30 antibody at a DAR of 16 (8 MMAE and 8 MMAF molecules per antibody). MMAE, being a substrate for multidrug resistance (MDR) transporters251,252, typically has diminished activity against MDR+ tumour cells. However, the hydrophobic nature of this toxin enables high levels of cell membrane permeability, leading to eradication of neighbouring cells via the bystander effect253,254. MMAF is not susceptible to drug export and is highly potent against MDR+ cells; however, this toxin is unlikely to have bystander effects owing to its limited cellular permeability254,255. Based on these distinct characteristics, the researchers hypothesized that simultaneous delivery of MMAE and MMAF would have enhanced and synergistic activities250. This dual-drug ADC demonstrated potent activity in a mouse xenograft model of CD30+ MDR-expressing anaplastic large-cell lymphoma (ALCL), resulting in complete eradication of cancer cells in three of five mice. By contrast, an MMAF-only ADC, with a DAR of 8, had less activity and eradicated all cancer cells in only one of five mice, and the equivalent MMAE-only ADC had no observable antitumour activity. In a mouse xenograft model of Hodgkin lymphoma that featured heterogeneous CD30 expression, both the dual-drug ADC and the MMAE-only ADC resulted in complete inhibition of tumour growth, probably attributable to bystander cell killing mediated by MMAE253,254. Conversely, the MMAF-only equivalent ADC conferred only modest delays in tumour growth250.
The above-mentioned pioneering work250 demonstrated the potential of dual-drug ADCs. However, the ability to adjust the DAR, and especially the production of low-DAR ADCs, is limited; homogeneous conjugation by cysteine alkylation relies on the ability to fully utilize all eight interchain-derived cysteine residues within a monoclonal antibody, resulting in an ADC with a fixed DAR of 16 (8 of each payload). To address this issue, researchers developed an enzymatic conjugation method that involves orthogonally functionalized linkers72 (Fig. 6c). Using microbial transglutaminase for site-specific linker installation71,95,96,97,98,256, these investigators produced highly homogeneous anti-HER2 dual-drug ADCs containing MMAE and MMAF with a range of defined DARs (MMAE to MMAF ratios of 2:2, 2:4 and 4:2). In vivo testing in two mouse xenograft models of aggressive HER2low breast cancer with moderate resistance to hydrophobic payloads such as MMAE demonstrated exceptional antitumour activity of the MMAE–MMAF 4:2 ADC. In a JIMT-1–MDA-MB-231 admixed orthotopic mouse model, a single 3 mg/kg dose of the 4:2 dual-drug ADC resulted in complete tumour eradication in all five mice, outperforming a single 3 mg/kg dose of the MMAE-only ADC (which resulted in complete tumour eradication in three of five mice) and a 1:1 cocktail of single-drug ADCs (MMAE-only plus MMAF-only ADCs both with a DAR of 4 administered at doses of 3 mg/kg, which resulted in complete tumour eradication in two of five mice). Antitumour activity of the dual-drug ADC was maintained even at the lower dose of 1 mg/kg. Similar results were observed in the HCC1954-TDR breast tumour model, which is known to be resistant to trastuzumab emtansine72,257. Biodistribution analysis using the HER2low admixed tumour model suggests that the use of two single-drug ADCs that target the same antigen can lead to binding competition and inefficient payload delivery. By contrast, other investigators have reported that co-administration of T-DM1 with trastuzumab improves the extent of tissue penetration of the ADC in a mouse HER2high NCI-N87 xenograft model by mitigating the binding-site barrier effect258. These findings indicate that dual-drug ADCs could be particularly advantageous for the treatment of patients with antigenlow tumours.
Beyond ADCs with MMAE and MMAF combination payloads, the clinical potential of the dual-drug ADC format has been explored further with the development of ADCs combining two different payload classes. Examples include the combination of the anti-microtubule agent hemiasterlin plus a TLR agonist conjugated to an anti-FolRα antibody, which has demonstrated synergistic antitumour activity and immunological memory in mouse models259. This study, along with those described previously72,250, highlights the clinical potential of dual-drug ADCs. However, not all studies testing dual-drug ADCs have demonstrated meaningful synergistic effects, particularly those involving different payload classes260,261,262. For example, investigators developed an anti-HER2 ADC equipped with MMAE plus SG3457, an ultrapotent PBD dimer capable of damaging DNA via crosslinking, at a DAR of 2 + 2 (ref. 260). Likewise, a HER2-targeted ADC equipped with MMAF and the highly potent topoisomerase II inhibitor PNU-15968, also at a DAR of 2 + 2 was developed261. Despite both of these ADCs being capable of exerting dual mechanisms of action, neither agent demonstrated an improvement in in vitro potency when compared with their corresponding single-drug ADCs. These findings highlight the importance of selecting payloads with appropriate mechanisms of action, ensuring balanced potency between the two payloads selected and optimizing DARs to achieve optimal therapeutic outcomes. Efforts to fully understand and maximize the potential of dual-drug ADCs are still in the early stages of exploration.
Conclusions
The therapeutic potential of ADCs is tremendous, yet realizing this potential will require several key challenges to be overcome such as drug resistance, intratumour and intertumour heterogeneity and the risks of TRAEs to be addressed. Emerging ADC modalities, including bispecific and dual-drug ADCs, show potential to address resistance and tumour heterogeneity, while PDCs could potentially increase tumour specificity and reduce the incidence of adverse events. Combining the ADC platform with other intervention strategies, such as immunomodulation and degradation of traditionally undruggable targets, provides opportunities to implement multimodal cancer treatment along with chemotherapy, radiotherapy, immunotherapy and other targeted therapy. However, the full potential of ADCs cannot be unleashed without comprehensive patient stratification and biomarker identification, which are often overlooked during the early stages of novel ADC development. Biomarkers are essential to identify patient populations that are most likely to derive benefit from a given ADC, thereby enabling personalized medicine. Given the heterogeneous nature of many solid tumours, reliable biomarkers are crucial for optimal patient selection. The need for biomarkers is particularly important when testing novel ADC modalities such as those discussed in this Review, owing to their increased complexity, multimodal nature and/or involvement in difficult-to-predict effects such as anticancer immunity. ADC development is on the brink of transformative growth, holding the promise to substantially alter the cancer treatment landscape. As we better understand tumour biology and improve ADC design, we will move closer to the goal of truly effective, safe and personalized cancer treatments, which will ultimately bring renewed hope for patients with intractable cancers.
References
Drago, J. Z., Modi, S. & Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021).
Dumontet, C., Reichert, J. M., Senter, P. D., Lambert, J. M. & Beck, A. Antibody–drug conjugates come of age in oncology. Nat. Rev. Drug. Discov. 22, 641–661 (2023).
Tarantino, P., Ricciuti, B., Pradhan, S. M. & Tolaney, S. M. Optimizing the safety of antibody–drug conjugates for patients with solid tumours. Nat. Rev. Clin. Oncol. 20, 558–576 (2023).
Beck, A., Goetsch, L., Dumontet, C. & Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug. Discov. 16, 315–337 (2017).
Maecker, H., Jonnalagadda, V., Bhakta, S., Jammalamadaka, V. & Junutula, J. R. Exploration of the antibody–drug conjugate clinical landscape. MAbs 15, 2229101 (2023).
Grand View Research. Antibody Drug Conjugates Market Size, Share & Trends Analysis Report by Application (Blood Cancer, Breast Cancer), by Technology ({Type-Cleavable, Non-Cleavable}), by Region, and Segment Forecasts, 2023–2030, https://www.grandviewresearch.com/industry-analysis/antibody-drug-conjugates-market (2024).
Dagogo-Jack, I. & Shaw, A. T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018).
Loganzo, F., Sung, M. & Gerber, H.-P. Mechanisms of resistance to antibody–drug conjugates. Mol. Cancer Ther. 15, 2825–2834 (2016).
García-Alonso, S., Ocaña, A. & Pandiella, A. Resistance to antibody–drug conjugates. Cancer Res. 78, 2159–2165 (2018).
Norsworthy, K. J. et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist 23, 1103–1108 (2018).
Ricart, A. D. Antibody–drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 17, 6417–6427 (2011).
Yu, J., Song, Y. & Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 13, 45 (2020).
Hock, M. B., Thudium, K. E., Carrasco-Triguero, M. & Schwabe, N. F. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 17, 35–43 (2015).
Gorovits, B. & Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody–drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 62, 217–223 (2013).
Yamazoe, S. et al. Impact of drug conjugation on thermal and metabolic stabilities of aglycosylated and N-glycosylated antibodies. Bioconjug. Chem. 33, 576–585 (2022).
Gutierrez, C. & Schiff, R. HER2: biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 135, 55–62 (2011).
Stepan, L. P. et al. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J. Histochem. Cytochem. 59, 701–710 (2011).
Pegram, M. D. et al. First-in-human, phase 1 dose-escalation study of biparatopic anti-HER2 antibody–drug conjugate MEDI4276 in patients with HER2-positive advanced breast or gastric cancer. Mol. Cancer Ther. 20, 1442–1453 (2021).
Le Joncour, V. et al. A novel anti-HER2 antibody-drug conjugate XMT-1522 for HER2-positive breast and gastric cancers resistant to trastuzumab emtansine. Mol. Cancer Ther. 18, 1721–1730 (2019).
Mersana Therapeutics. Mersana Therapeutics announces partial clinical hold for XMT-1522 clinical trial. https://ir.mersana.com/news-releases/news-release-details/mersana-therapeutics-announces-partial-clinical-hold-xmt-1522 (2018).
Duvall, J. R. et al. XMT-2056, a HER2-targeted immunosynthen STING-agonist antibody-drug conjugate, binds a novel epitope of HER2 and shows increased anti-tumor activity in combination with trastuzumab and pertuzumab. Cancer Res. 82 (Suppl. 12), Abstr. 3503 (2022).
Mersana Therapeutics. Mersana Therapeutics announces clinical hold on XMT-2056 phase 1 clinical trial. https://ir.mersana.com/news-releases/news-release-details/mersana-therapeutics-announces-clinical-hold-xmt-2056-phase-1 (2023).
King, G. T. et al. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Invest. New Drugs 36, 836–847 (2018).
Gan, H. K., Cvrljevic, A. N. & Johns, T. G. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 280, 5350–5370 (2013).
Phillips, A. C. et al. ABT-414, an antibody–drug conjugate targeting a tumor-selective EGFR epitope. Mol. Cancer Ther. 15, 661–669 (2016).
Trerotola, M. et al. Trop-2 cleavage by ADAM10 is an activator switch for cancer growth and metastasis. Neoplasia 23, 415–428 (2021).
Alberti, S., Trerotola, M. & Guerra, E. The Hu2G10 tumor-selective anti-Trop-2 monoclonal antibody targets the cleaved-activated Trop-2 and shows therapeutic efficacy against multiple human cancers. Cancer Res. 82 (Suppl. 12), Abstr. 340 (2022).
Kim, H. et al. LCB84, a TROP2-targeted ADC, for treatment of solid tumors that express TROP-2 using the hu2G10 tumor-selective anti-TROP2 monoclonal antibody, a proprietary site-directed conjugation technology and plasma-stable tumor-selective linker chemistry. Cancer Res. 82 (Suppl. 12), Abstr. 328 (2022).
Li, C.-W. et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187–201.e10 (2018).
de Goeij, B. E. C. G. et al. High turnover of tissue factor enables efficient intracellular delivery of antibody–drug conjugates. Mol. Cancer Ther. 14, 1130–1140 (2015).
Thurber, G. M., Schmidt, M. M. & Wittrup, K. D. Antibody tumor penetration: transport opposed by systemic and antigen-mediated clearance. Adv. Drug. Deliv. Rev. 60, 1421–1434 (2008).
Adams, G. P. et al. High affinity restricts the localization and tumor penetration of single-chain Fv antibody molecules. Cancer Res. 61, 4750–4755 (2001).
Samantasinghar, A. et al. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. Biomed. Pharmacother. 161, 114408 (2023).
Yadav, A., Mandal, M. K. & Dubey, K. K. In vitro cytotoxicity study of cyclophosphamide, etoposide and paclitaxel on monocyte macrophage cell line raw 264.7. Indian J. Microbiol. 60, 511–517 (2020).
Tumey, L. N. et al. Optimization of tubulysin antibody–drug conjugates: a case study in addressing ADC metabolism. ACS Med. Chem. Lett. 7, 977–982 (2016).
Staben, L. R. et al. Stabilizing a tubulysin antibody–drug conjugate to enable activity against multidrug-resistant tumors. ACS Med. Chem. Lett. 8, 1037–1041 (2017).
Burke, P. J. et al. Glucuronide-linked antibody–tubulysin conjugates display activity in MDR+ and heterogeneous tumor models. Mol. Cancer Ther. 17, 1752–1760 (2018).
Hamilton, J. Z. et al. Improving antibody–tubulysin conjugates through linker chemistry and site-specific conjugation. ChemMedChem 16, 1077–1081 (2021).
Rottey, S. et al. Phase I/IIa trial of BMS-986148, an anti-mesothelin antibody–drug conjugate, alone or in combination with nivolumab in patients with advanced solid tumors. Clin. Cancer Res. 28, 95–105 (2022).
Yao, H.-P., Zhao, H., Hudson, R., Tong, X.-M. & Wang, M.-H. Duocarmycin-based antibody–drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: pharmaceutical strategy and clinical progress. Drug Discov. Today 26, 1857–1874 (2021).
Yu, S.-F. et al. A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin. Cancer Res. 21, 3298–3306 (2015).
Stefan, N. et al. Highly potent, anthracycline-based antibody-drug conjugates generated by enzymatic, site-specific conjugation. Mol. Cancer Ther. 16, 879–892 (2017).
Holte, D. et al. Evaluation of PNU-159682 antibody drug conjugates (ADCs). Bioorg. Med. Chem. Lett. 30, 127640 (2020).
Pahl, A., Lutz, C. & Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 30, 85–89 (2018).
Liu, Y. et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520, 697–701 (2015).
Vaisitti, T. et al. Anti-CD37 α-amanitin-conjugated antibodies as potential therapeutic weapons for Richter syndrome. Blood 140, 1565–1569 (2022).
Ackerman, S. E. et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat. Cancer 2, 18–33 (2021).
Hong, K. B. & An, H. Degrader–antibody conjugates: emerging new modality. J. Med. Chem. 66, 140–148 (2023).
Khera, E. et al. Cellular-resolution imaging of bystander payload tissue penetration from antibody–drug conjugates. Mol. Cancer Ther. 21, 310–321 (2022).
Staudacher, A. H. & Brown, M. P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer 117, 1736–1742 (2017).
Dei, S., Braconi, L., Romanelli, M. N. & Teodori, E. Recent advances in the search of BCRP- and dual P-gp/BCRP-based multidrug resistance modulators. Cancer Drug Resist. 2, 710–743 (2019).
Buecheler, J. W., Winzer, M., Tonillo, J., Weber, C. & Gieseler, H. Impact of payload hydrophobicity on the stability of antibody–drug conjugates. Mol. Pharm. 15, 2656–2664 (2018).
Lyon, R. P. et al. Reducing hydrophobicity of homogeneous antibody–drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 33, 733–735 (2015).
Lundahl, M. L. E., Fogli, S., Colavita, P. E. & Scanlan, E. M. Aggregation of protein therapeutics enhances their immunogenicity: causes and mitigation strategies. RSC Chem. Biol. 2, 1004–1020 (2021).
Lyski, R. D. et al. Development of novel antibody–camptothecin conjugates. Mol. Cancer Ther. 20, 329–339 (2021).
Simmons, J. K., Burke, P. J., Cochran, J. H., Pittman, P. G. & Lyon, R. P. Reducing the antigen-independent toxicity of antibody–drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicol. Appl. Pharmacol. 392, 114932 (2020).
Meyer, D. W. et al. An in vitro assay using cultured Kupffer cells can predict the impact of drug conjugation on in vivo antibody pharmacokinetics. Mol. Pharm. 17, 802–809 (2020).
Zhao, H. et al. Modulation of macropinocytosis-mediated internalization decreases ocular toxicity of antibody–drug conjugates. Cancer Res. 78, 2115–2126 (2018).
Zhao, H. et al. Inhibition of megakaryocyte differentiation by antibody–drug conjugates (ADCs) is mediated by macropinocytosis: implications for ADC-induced thrombocytopenia. Mol. Cancer Ther. 16, 1877–1886 (2017).
Guffroy, M. et al. Liver microvascular injury and thrombocytopenia of antibody–calicheamicin conjugates in cynomolgus monkeys—mechanism and monitoring. Clin. Cancer Res. 23, 1760–1770 (2017).
Burke, P. J. et al. Optimization of a PEGylated glucuronide-monomethylauristatin E linker for antibody-drug conjugates. Mol. Cancer Ther. 16, 116–123 (2017).
Ochtrop, P. et al. Compact hydrophilic electrophiles enable highly efficacious high DAR ADCs with excellent in vivo PK profile. Chem. Sci. 14, 2259–2266 (2023).
Viricel, W. et al. Monodisperse polysarcosine-based highly-loaded antibody–drug conjugates. Chem. Sci. 10, 4048–4053 (2019).
Hu, Y., Hou, Y., Wang, H. & Lu, H. Polysarcosine as an alternative to PEG for therapeutic protein conjugation. Bioconjug. Chem. 29, 2232–2238 (2018).
Conilh, L. et al. Exatecan antibody drug conjugates based on a hydrophilic polysarcosine drug-linker platform. Pharmaceuticals 14, 247 (2021).
Weng, W. et al. Antibody–exatecan conjugates with a novel self-immolative moiety overcome resistance in colon and lung cancer. Cancer Discov. 13, 950–973 (2023).
Tsuchikama, K. & An, Z. Antibody–drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 9, 33–46 (2018).
Su, D. & Zhang, D. Linker design impacts antibody–drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front. Pharmacol. 12, 687926 (2021).
Baah, S., Laws, M. & Rahman, K. M. Antibody–drug conjugates—a tutorial review. Molecules 26, 2943 (2021).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Anami, Y. et al. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 9, 2512 (2018).
Yamazaki, C. M. et al. Antibody–drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 12, 3528 (2021).
Ha, S. Y. Y. et al. An enzymatically cleavable tripeptide linker for maximizing the therapeutic index of antibody–drug conjugates. Mol. Cancer Ther. 21, 1449–1461 (2022).
Jeffrey, S. C. et al. Development and properties of beta-glucuronide linkers for monoclonal antibody–drug conjugates. Bioconjug. Chem. 17, 831–840 (2006).
Chuprakov, S. et al. Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody–drug conjugates. Bioconjug. Chem. 32, 746–754 (2021).
Bargh, J. D. et al. Sulfatase-cleavable linkers for antibody–drug conjugates. Chem. Sci. 11, 2375–2380 (2020).
Kern, J. C. et al. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J. Am. Chem. Soc. 138, 1430–1445 (2016).
Lerchen, H.-G. et al. Tailored linker chemistries for the efficient and selective activation of ADCs with KSPi payloads. Bioconjug. Chem. 31, 1893–1898 (2020).
Miller, J. T., Vitro, C. N., Fang, S., Benjamin, S. R. & Tumey, L. N. Enzyme-agnostic lysosomal screen identifies new legumain-cleavable ADC linkers. Bioconjug. Chem. 32, 842–858 (2021).
Junutula, J. R. et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 26, 925–932 (2008).
Axup, J. Y. et al. Synthesis of site-specific antibody–drug conjugates using unnatural amino acids. Proc. Natl Acad. Sci. USA 109, 16101–16106 (2012).
Zimmerman, E. S. et al. Production of site-specific antibody–drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjug. Chem. 25, 351–361 (2014).
VanBrunt, M. P. et al. Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody–drug conjugates using click cycloaddition chemistry. Bioconjug. Chem. 26, 2249–2260 (2015).
Bryden, F. et al. Regioselective and stoichiometrically controlled conjugation of photodynamic sensitizers to a HER2 targeting antibody fragment. Bioconjug. Chem. 25, 611–617 (2014).
Schumacher, F. F. et al. Next generation maleimides enable the controlled assembly of antibody–drug conjugates via native disulfide bond bridging. Org. Biomol. Chem. 12, 7261–7269 (2014).
Behrens, C. R. et al. Antibody–drug conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol. Pharm. 12, 3986–3998 (2015).
Maruani, A. et al. A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat. Commun. 6, 6645 (2015).
Bahou, C. et al. Highly homogeneous antibody modification through optimisation of the synthesis and conjugation of functionalised dibromopyridazinediones. Org. Biomol. Chem. 16, 1359–1366 (2018).
Forte, N., Chudasama, V. & Baker, J. R. Homogeneous antibody–drug conjugates via site-selective disulfide bridging. Drug Discov. Today Technol. 30, 11–20 (2018).
Fujii, T. et al. AJICAP second generation: improved chemical site-specific conjugation technology for antibody–drug conjugate production. Bioconjug. Chem. 34, 728–738 (2023).
Zhou, Q. et al. Site-specific antibody–drug conjugation through glycoengineering. Bioconjug. Chem. 25, 510–520 (2014).
van Geel, R. et al. Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody-drug conjugates. Bioconjug. Chem. 26, 2233–2242 (2015).
Manabe, S. et al. Characterization of antibody products obtained through enzymatic and nonenzymatic glycosylation reactions with a glycan oxazoline and preparation of a homogeneous antibody-drug conjugate via Fc N-glycan. Bioconjug. Chem. 30, 1343–1355 (2019).
de Bever, L. et al. Generation of DAR1 antibody–drug conjugates for ultrapotent payloads using tailored GlycoConnect technology. Bioconjug. Chem. 34, 538–548 (2023).
Jeger, S. et al. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 49, 9995–9997 (2010).
Strop, P. et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 20, 161–167 (2013).
Dennler, P. et al. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjug. Chem. 25, 569–578 (2014).
Anami, Y. et al. Enzymatic conjugation using branched linkers for constructing homogeneous antibody–drug conjugates with high potency. Org. Biomol. Chem. 15, 5635–5642 (2017).
Rabuka, D., Rush, J. S., deHart, G. W., Wu, P. & Bertozzi, C. R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 7, 1052–1067 (2012).
Drake, P. M. et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 25, 1331–1341 (2014).
Beerli, R. R., Hell, T., Merkel, A. S. & Grawunder, U. Sortase enzyme-mediated generation of site-specifically conjugated antibody drug conjugates with high in vitro and in vivo potency. PLoS ONE 10, e0131177 (2015).
Pan, L. et al. Sortase A-generated highly potent anti-CD20–MMAE conjugates for efficient elimination of B-lineage lymphomas. Small 13, 1602267 (2017).
Antos, J. M. et al. Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci. 89, 15.3.1–15.3.19 (2017).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today 20, 838–847 (2015).
Beishenaliev, A. et al. Bispecific antibodies for targeted delivery of anti-cancer therapeutic agents: a review. J. Control. Release 359, 268–286 (2023).
Kantarjian, H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376, 836–847 (2017).
Linke, R., Klein, A. & Seimetz, D. Catumaxomab: clinical development and future directions. MAbs 2, 129–136 (2010).
Maruani, A. Bispecifics and antibody–drug conjugates: a positive synergy. Drug Discov. Today Technol. 30, 55–61 (2018).
Friedman, L. M. et al. Synergistic down-regulation of receptor tyrosine kinases by combinations of mAbs: implications for cancer immunotherapy. Proc. Natl Acad. Sci. USA 102, 1915–1920 (2005).
Ben-Kasus, T., Schechter, B., Lavi, S., Yarden, Y. & Sela, M. Persistent elimination of ErbB-2/HER2-overexpressing tumors using combinations of monoclonal antibodies: relevance of receptor endocytosis. Proc. Natl Acad. Sci. USA 106, 3294–3299 (2009).
Li, J. Y. et al. A biparatopic HER2-targeting antibody–drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 29, 117–129 (2016).
Toader, D. et al. Structure–cytotoxicity relationships of analogues of N14-desacetoxytubulysin H. J. Med. Chem. 59, 10781–10787 (2016).
Oganesyan, V. et al. Structural insights into the mechanism of action of a biparatopic anti-HER2 antibody. J. Biol. Chem. 293, 8439–8448 (2018).
Modi, S. et al. Antitumor activity and safety of trastuzumab deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a phase Ib study. J. Clin. Oncol. 38, 1887–1896 (2020).
Hamblett, K. J. et al. ZW49, a HER2 targeted biparatopic antibody drug conjugate for the treatment of HER2 expressing cancers. Cancer Res. 79 (Suppl. 4), Abstr. P6-17-13 (2019).
Barnscher, S. D., Rojas, A. H., Hamblett, K. J. & Escalante, N. Zanidatamab zovodotin (ZW49) induces hallmarks of immunogenic cell death and is active in patient-derived xenograft models of gastric cancer. Cancer Res. 83 (Suppl. 7), Abstr. 2633 (2023).
Hamblett, K. et al. Anti-her2 biparatopic antibody–drug conjugates and methods of use. US Patent US20210346508A1 (2021).
Weisser, N. E. et al. An anti-HER2 biparatopic antibody that induces unique HER2 clustering and complement-dependent cytotoxicity. Nat. Commun. 14, 1394 (2023).
Jhaveri, K. et al. 460MO preliminary results from a phase I study using the bispecific, human epidermal growth factor 2 (HER2)-targeting antibody-drug conjugate (ADC) zanidatamab zovodotin (ZW49) in solid cancers. Ann. Oncol. 33, S749–S750 (2022).
Katz, J., Janik, J. E. & Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 17, 6428–6436 (2011).
Palanca-Wessels, M. C. A. et al. Safety and activity of the anti-CD79B antibody-drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study. Lancet Oncol. 16, 704–715 (2015).
Alt, M., Stecca, C., Tobin, S., Jiang, D. M. & Sridhar, S. S. Enfortumab vedotin in urothelial cancer. Ther. Adv. Urol. 12, 1756287220980192 (2020).
DaSilva, J. O. et al. A biparatopic antibody–drug conjugate to treat MET-expressing cancers, including those that are unresponsive to MET pathway blockade. Mol. Cancer Ther. 20, 1966–1976 (2021).
Perez Bay, A. E. et al. A bispecific METxMET antibody-drug conjugate with cleavable linker is processed in recycling and late endosomes. Mol. Cancer Ther. 22, 357–370 (2023).
DaSilva, J. O. et al. A biparatopic antibody that modulates MET trafficking exhibits enhanced efficacy compared with parental antibodies in MET-driven tumor models. Clin. Cancer Res. 26, 1408–1419 (2020).
Drilon, A. E. et al. A phase 1/2 study of REGN5093-M114, a METxMET antibody–drug conjugate, in patients with mesenchymal epithelial transition factor (MET)-overexpressing NSCLC. J. Clin. Oncol. 40, TPS8593 (2022).
Gomatou, G., Syrigos, N. & Kotteas, E. Osimertinib resistance: molecular mechanisms and emerging treatment options. Cancers 15, 841 (2023).
Cho, B. C. Dr Cho on first-line amivantamab plus lazertinib in EGFR-mutant NSCLC. OncLive https://www.onclive.com/view/dr-cho-on-the-mariposa-trial-of-first-line-amivantamab-plus-lazertinib-in-egfr-mutant-nsclc (2023).
Sellmann, C. et al. Balancing selectivity and efficacy of bispecific epidermal growth factor receptor (EGFR) × c-MET antibodies and antibody–drug conjugates. J. Biol. Chem. 291, 25106–25119 (2016).
Comer, F. et al. AZD9592: an EGFR-cMET bispecific antibody-drug conjugate (ADC) targeting key oncogenic drivers in non-small-cell lung cancer (NSCLC) and beyond. Cancer Res. 83 (Suppl. 7), Abstr. 5736 (2023).
McGrath, L. et al. Evaluation of the relationship between target expression and in vivo anti-tumor efficacy of AZD9592, an EGFR/c-MET targeted bispecific antibody drug conjugate. Cancer Res. 83 (Suppl. 7), Abstr. 5737 (2023).
Knuehl, C. et al. M1231 is a bispecific anti-MUC1xEGFR antibody-drug conjugate designed to treat solid tumors with MUC1 and EGFR co-expression. Cancer Res. 82 (Suppl. 12), Abstr. 5284 (2022).
Wan, W. et al. BL-B01D1, a novel EGFR×HER3-targeting ADC, demonstrates robust anti-tumor efficacy in preclinical evaluation. Cancer Res. 83 (Suppl. 7), Abstr. 2642 (2023).
Sorkin, A. & Goh, L. K. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 315, 683–696 (2009).
Austin, C. D. et al. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 15, 5268–5282 (2004).
DeVay, R. M., Shelton, D. L. & Liang, H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J. Biol. Chem. 288, 10805–10818 (2013).
DeVay, R. M., Yamamoto, L., Shelton, D. L. & Liang, H. Common proprotein convertase subtilisin/kexin type 9 (PCSK9) epitopes mediate multiple routes for internalization and function. PLoS ONE 10, e0125127 (2015).
Tuli, A. et al. Amyloid precursor-like protein 2 increases the endocytosis, instability, and turnover of the H2-K(d) MHC class I molecule. J. Immunol. 181, 1978–1987 (2008).
Tuli, A. et al. Mechanism for amyloid precursor-like protein 2 enhancement of major histocompatibility complex class I molecule degradation. J. Biol. Chem. 284, 34296–34307 (2009).
Andreev, J. et al. Bispecific antibodies and antibody-drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol. Cancer Ther. 16, 681–693 (2017).
DeVay, R. M. et al. Improved lysosomal trafficking can modulate the potency of antibody drug conjugates. Bioconjug. Chem. 28, 1102–1114 (2017).
Merchant, M. et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl Acad. Sci. USA 110, E2987–E2996 (2013).
Liu, L. et al. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. 20, 6059–6070 (2014).
Oh, Y. M. et al. A new anti-c-Met antibody selected by a mechanism-based dual-screening method: therapeutic potential in cancer. Mol. Cell 34, 523–529 (2012).
Neijssen, J. et al. Discovery of amivantamab (JNJ-61186372), a bispecific antibody targeting EGFR and MET. J. Biol. Chem. 296, 100641 (2021).
Kim, S.-B. et al. First-in-human phase I study of aprutumab ixadotin, a fibroblast growth factor receptor 2 antibody–drug conjugate (BAY 1187982) in patients with advanced cancer. Target. Oncol. 14, 591–601 (2019).
Lemech, C. et al. A first-in-human, phase 1, dose-escalation study of ABBV-176, an antibody–drug conjugate targeting the prolactin receptor, in patients with advanced solid tumors. Invest. New Drugs 38, 1815–1825 (2020).
Zhao, P. et al. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 10, 1589–1600 (2020).
Rautio, J. et al. Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 7, 255–270 (2008).
Abet, V., Filace, F., Recio, J., Alvarez-Builla, J. & Burgos, C. Prodrug approach: an overview of recent cases. Eur. J. Med. Chem. 127, 810–827 (2017).
Rautio, J., Meanwell, N. A., Di, L. & Hageman, M. J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 17, 559–587 (2018).
Erster, O. et al. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J. Control. Release 161, 804–812 (2012).
Desnoyers, L. R. et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 5, 207ra144 (2013).
Geiger, M. et al. Protease-activation using anti-idiotypic masks enables tumor specificity of a folate receptor 1-T cell bispecific antibody. Nat. Commun. 11, 3196 (2020).
Sulea, T. et al. Structure-based engineering of pH-dependent antibody binding for selective targeting of solid-tumor microenvironment. MAbs 12, 1682866 (2020).
Lucchi, R., Bentanachs, J. & Oller-Salvia, B. The masking game: design of activatable antibodies and mimetics for selective therapeutics and cell control. ACS Cent. Sci. 7, 724–738 (2021).
Liu, Y., Nguyen, A. W. & Maynard, J. A. Engineering antibodies for conditional activity in the solid tumor microenvironment. Curr. Opin. Biotechnol. 78, 102809 (2022).
Bleuez, C., Koch, W. F., Urbach, C., Hollfelder, F. & Jermutus, L. Exploiting protease activation for therapy. Drug Discov. Today 27, 1743–1754 (2022).
Liu, X. et al. A cross-reactive pH-dependent EGFR antibody with improved tumor selectivity and penetration obtained by structure-guided engineering. Mol. Ther. Oncolytics 27, 256–269 (2022).
Ulisse, S., Baldini, E., Sorrenti, S. & D’Armiento, M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr. Cancer Drug Targets 9, 32–71 (2009).
Uhland, K. Matriptase and its putative role in cancer. Cell. Mol. Life Sci. 63, 2968–2978 (2006).
LeBeau, A. M. et al. Imaging a functional tumorigenic biomarker in the transformed epithelium. Proc. Natl Acad. Sci. USA 110, 93–98 (2013).
Liu, C., Sun, C., Huang, H., Janda, K. & Edgington, T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 63, 2957–2964 (2003).
Singh, S. et al. Nonclinical efficacy and safety of CX-2029, an anti-CD71 probody–drug conjugate. Mol. Cancer Ther. 21, 1326–1336 (2022).
CytomX Therapeutics Inc. CytomX therapeutics presents overview of conditionally-activated antibody-drug conjugate (ADC) programs including next generation EpCAM-targeting CX-2051. CytomX Press Release Details https://ir.cytomx.com/news-releases/news-release-details/cytomx-therapeutics-presents-overview-conditionally-activated/ (2022).
Chomet, M. et al. The tumor targeting performance of anti-CD166 Probody drug conjugate CX-2009 and its parental derivatives as monitored by 89Zr-immuno-PET in xenograft bearing mice. Theranostics 10, 5815–5828 (2020).
Johnson, M. et al. Phase I, first-in-human study of the probody therapeutic CX-2029 in adults with advanced solid tumor malignancies. Clin. Cancer Res. 27, 4521–4530 (2021).
Boni, V. et al. Praluzatamab ravtansine, a CD166-targeting antibody–drug conjugate, in patients with advanced solid tumors: an open-label phase I/II trial. Clin. Cancer Res. 28, 2020–2029 (2022).
Swart, G. W. M. Activated leukocyte cell adhesion molecule (CD166/ALCAM): developmental and mechanistic aspects of cell clustering and cell migration. Eur. J. Cell Biol. 81, 313–321 (2002).
Garcia-Corbacho, J. et al. PROCLAIM-CX-2009: a first-in-human trial to evaluate CX-2009 in adults with metastatic or locally advanced unresectable solid tumors. Ann. Oncol. 28, v140 (2017).
Trang, V. H. et al. A coiled-coil masking domain for selective activation of therapeutic antibodies. Nat. Biotechnol. 37, 761–765 (2019).
Huang, L. et al. Preclinical evaluation of a next-generation, EGFR targeting ADC that promotes regression in KRAS or BRAF mutant tumors. Cancer Res. 76 (Suppl. 14), Abstr. 1217 (2016).
Bahn, J. D. et al. HTI-1511, a novel anti-EGFR-ADC, overcomes mutation resistance and demonstrates significant activity against multiple tumor types in preclinical studies. Cancer Res. 77 (Suppl. 13), Abstr. 50 (2017).
Kang, J. C. et al. Engineering a HER2-specific antibody-drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol. 37, 523–526 (2019).
Sharp, L. L. et al. Anti-tumor efficacy of BA3011, a novel conditionally active biologic (CAB) anti-AXL-ADC. Cancer Res. 78 (Suppl. 13), Abstr. 827 (2018).
Sharp, L. L. et al. Anti-tumor efficacy of BA3021, a novel conditionally active biologic (CAB) anti-ROR2 ADC. Cancer Res. 78 (Suppl. 13), Abstr. 833 (2018).
Khan, M. A. G. Halozyme terminates license agreement with Abzena. https://www.spglobal.com/marketintelligence/en/news-insights/blog/essential-ir-insights-newsletter-fall-2023 (2018).
Chang, H. W. et al. Generating tumor-selective conditionally active biologic anti-CTLA4 antibodies via protein-associated chemical switches. Proc. Natl Acad. Sci. USA 118, e2020606118 (2021).
BioAtla. CAB Portfolio. BioAtla https://www.bioatla.com/cab-portfolio/ (2016).
Seidel, J. A., Otsuka, A. & Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front. Oncol. 8, 86 (2018).
Urban-Wojciuk, Z. et al. The role of TLRs in anti-cancer immunity and tumor rejection. Front. Immunol. 10, 2388 (2019).
Kaczanowska, S., Joseph, A. M. & Davila, E. TLR agonists: our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 93, 847–863 (2013).
Le Naour, J. & Kroemer, G. Trial watch: toll-like receptor ligands in cancer therapy. Oncoimmunology 12, 2180237 (2023).
Amouzegar, A., Chelvanambi, M., Filderman, J. N., Storkus, W. J. & Luke, J. J. STING agonists as cancer therapeutics. Cancers 13, 2695 (2021).
Su, T. et al. STING activation in cancer immunotherapy. Theranostics 9, 7759–7771 (2019).
Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 89, 107087 (2020).
Hamid, O., Ismail, R. & Puzanov, I. Intratumoral immunotherapy — update 2019. Oncologist 25, e423–e438 (2019).
Rodell, C. B. et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2, 578–588 (2018).
Huang, L., Ge, X., Liu, Y., Li, H. & Zhang, Z. The role of toll-like receptor agonists and their nanomedicines for tumor immunotherapy. Pharmaceutics 14, 1228 (2022).
Gadd, A. J. R., Greco, F., Cobb, A. J. A. & Edwards, A. D. Targeted activation of toll-like receptors: conjugation of a toll-like receptor 7 agonist to a monoclonal antibody maintains antigen binding and specificity. Bioconjug. Chem. 26, 1743–1752 (2015).
Silverback Therapeutics Updates Strategic Priorities and Reports Fourth Quarter and Full Year 2021 Financial Results. https://www.businesswire.com/news/home/20220331005874/en/Silverback-Therapeutics-Updates-Strategic-Priorities-and-Reports-Fourth-Quarter-and-Full-Year-2021-Financial-Results (2022).
Doctor, V. Silverback Halts Oncology Programs, lays off 27% of staff. BioSpace https://www.biospace.com/article/silverback-halts-oncology-programs-lays-off-27-percent-of-staff/ (2022).
Janku, F. et al. 378 A first in-human, multicenter, open-label, dose-finding phase 1 study of the immune stimulator antibody conjugate NJH395 in patients with nonbreast HER2+ advanced malignancies. J. Immunother. Cancer 8 (Suppl. 3), A230 (2020).
Janku, F. et al. Preclinical characterization and phase I study of an anti-HER2-TLR7 immune-stimulator antibody conjugate in patients with HER2+ malignancies. Cancer Immunol. Res. 10, 1441–1461 (2022).
Ackerman, S. E., Alonso, M. N., Jackson, D. Y., Lee, A. & Engleman, E. G. Immunoconjugates targeting HER2. World Patent WO2020190725A1 (2020).
Bolt Biotherapeutics. Bolt biotherapeutics initiates phase 2 clinical studies of BDC-1001 in patients with HER2-positive cancer. https://investors.boltbio.com/news-releases/news-release-details/bolt-biotherapeutics-initiates-phase-2-clinical-studies-bdc-1001 (2023).
Li, B. T. et al. A phase 1/2 study of a first-in-human immune-stimulating antibody conjugate (ISAC) BDC-1001 in patients with advanced HER2-expressing solid tumors. J. Clin. Oncol. 41, 2538–2538 (2023).
Blum, L. K. et al. The CEA-targeted ISAC, BDC-2034, shows preclinical efficacy associated with innate immune activation, phagocytosis, and myeloid reprogramming. Cancer Res. 82 (Suppl. 12), Abstr. 2911 (2022).
Kenkel, J. A. et al. PD-L1-targeted ISAC combines myeloid cell activation, immune-checkpoint inhibition and ADCP to improve anti-tumor efficacy over anti-PD-L1 antibodies in preclinical models. Cancer Res. 82 (Suppl. 12), Abstr. 4252 (2022).
He, L. et al. Immune modulating antibody-drug conjugate (IM-ADC) for cancer immunotherapy. J. Med. Chem. 64, 15716–15726 (2021).
Fang, S. et al. Design and characterization of immune-stimulating imidazo[4,5-c]quinoline antibody-drug conjugates. Mol. Pharm. 19, 3228–3241 (2022).
Kuo, T. C. et al. TAC-001, a toll-like receptor 9 (TLR9) agonist antibody conjugate targeting B cells, promotes anti-tumor immunity and favorable safety profile following systemic administration in preclinical models. Cancer Res. 81 (Suppl. 13), Abstr. 1721 (2021).
Pons, J. et al. Transglutaminase-mediated conjugation. US Patent US20230130194A1 (2023).
Perez, C. et al. INCLINE-101, a phase 1/2, open label, dose escalation and expansion study of TAC-001 (a TLR9 agonist conjugated to a CD22 antibody) in patients with select advanced or metastatic solid tumors. J. Immunother. Cancer 10 (Suppl. 2), A788 (2022).
Zhu, Y. et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019).
Woo, S.-R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Woo, S.-R., Corrales, L. & Gajewski, T. F. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 36, 250–256 (2015).
Ohkuri, T. et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2, 1199–1208 (2014).
Demaria, O. et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl Acad. Sci. USA 112, 15408–15413 (2015).
Wu, Y.-T. et al. Tumor-targeted delivery of a STING agonist improvescancer immunotherapy. Proc. Natl Acad. Sci. USA 119, e2214278119 (2022).
Bukhalid, R. A. et al. Systemic administration of STING agonist antibody-drug conjugates elicit potent anti-tumor immune responses with minimal induction of circulating cytokines. Cancer Res. 80 (Suppl. 16), Abstr. 6706 (2020).
Duvall, J. R. et al. XMT-2056, a well-tolerated, Immunosynthen-based STING-agonist antibody-drug conjugate which induces anti-tumor immune activity. Cancer Res. 81 (Suppl. 13), Abstr. 1738 (2021).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018).
Mersana Therapeutics. Mersana Therapeutics announces FDA has lifted clinical hold on phase 1 clinical trial of XMT-2056. Mersana Therapeutics https://ir.mersana.com/news-releases/news-release-details/mersana-therapeutics-announces-fda-has-lifted-clinical-hold (2023).
Konstantinidou, M. et al. PROTACs – a game-changing technology. Expert Opin. Drug Discov. 14, 1255–1268 (2019).
Kelm, J. M. et al. PROTAC’ing oncoproteins: targeted protein degradation for cancer therapy. Mol. Cancer 22, 62 (2023).
An, S. & Fu, L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36, 553–562 (2018).
Liu, Z. et al. An overview of PROTACs: a promising drug discovery paradigm. Mol. Biomed. 3, 46 (2022).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Shorstova, T., Foulkes, W. D. & Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 124, 1478–1490 (2021).
Trojer, P. & Targeting, B. E. T. Bromodomains in cancer. Annu. Rev. Cancer Biol. 6, 313–336 (2022).
Liu, Z. et al. Drug discovery targeting bromodomain-containing protein 4. J. Med. Chem. 60, 4533–4558 (2017).
Pillow, T. H. et al. Antibody conjugation of a chimeric BET degrader enables in vivo activity. ChemMedChem 15, 17–25 (2020).
Min, J.-H. et al. Structure of an HIF-1alpha–pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886–1889 (2002).
Hon, W.-C. et al. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 417, 975–978 (2002).
Maneiro, M. A. et al. Antibody–PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol. 15, 1306–1312 (2020).
Cardillo, T. M., Govindan, S. V., Sharkey, R. M., Trisal, P. & Goldenberg, D. M. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: preclinical studies in human cancer xenograft models and monkeys. Clin. Cancer Res. 17, 3157–3169 (2011).
Goldenberg, D. M., Cardillo, T. M., Govindan, S. V., Rossi, E. A. & Sharkey, R. M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 6, 22496–22512 (2015).
Cardillo, T. M. et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2/SN-38 antibody-drug conjugate: characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug. Chem. 26, 919–931 (2015).
Dragovich, P. S. et al. Antibody-mediated delivery of chimeric BRD4 degraders. Part 1: exploration of antibody linker, payload loading, and payload molecular properties. J. Med. Chem. 64, 2534–2575 (2021).
Dragovich, P. S. et al. Antibody-mediated delivery of chimeric BRD4 degraders. Part 2: improvement of in vitro antiproliferation activity and in vivo antitumor efficacy. J. Med. Chem. 64, 2576–2607 (2021).
Chuang, S.-H. et al. Antibody protac conjugates. US Patent US20210015942A1 (2021).
Dragovich, P. S. et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorg. Med. Chem. Lett. 30, 126907 (2020).
Thompson, P. A., Edris, B., Coburn, C. A. & Baum, P. R. Antibody construct conjugates. World Patent WO2018227023A1 (2018).
Dragovich, P. S., Baker Dockrey, S. A., Pillow, T. H. & Zhang, D. Antibody-conjugated chemical inducers of degradation of brm and methods thereof. World Patent WO2022020288A1 (2022).
Palacino, J. et al. ORM-5029: a first-in-class targeted protein degradation therapy using antibody neodegrader conjugate (AnDC) for HER2-expressing breast cancer. Cancer Res. 82 (Suppl. 12), Abstr. 3933 (2022).
Saini, S. et al. Development of RNAscope multiplex-based assay for exploratory pharmacodynamic biomarkers assessment in breast cancer patients from phase I clinical trial of ORM-5029, a potent GSPT1 degrader. Cancer Res. 83 (Suppl. 7), Abstr. 2118 (2023).
Palacino, J. et al. ORM-6151: a first-in-class, anti-CD33 antibody-enabled GSPT1 degrader for AML. Blood 140, 3061–3062 (2022).
Palacino, J. et al. ORM-6151: a first-in-class CD33-antibody enabled GSPT1 degrader for AML. Cancer Res. 83 (Suppl. 7), Abstr. 2700 (2023).
Fishkin, N. & Park, P. U. Neodegrader conjugates. World Patent WO2021198965A1 (2021).
Matyskiela, M. E. et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257 (2016).
Hughes, S. J. & Ciulli, A. Molecular recognition of ternary complexes: a new dimension in the structure-guided design of chemical degraders. Essays Biochem. 61, 505–516 (2017).
Ju, Y. et al. ORM-5029: discovery of an antibody drug conjugate with first-in-class molecular glue degrader warhead for treatment of HER2-positive breast cancer | Poster Board #3753. https://acs.digitellinc.com/sessions/584447/view (2023).
Cotton, A. D., Nguyen, D. P., Gramespacher, J. A., Seiple, I. B. & Wells, J. A. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 143, 593–598 (2021).
Marei, H. et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature 610, 182–189 (2022).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Ahn, G. et al. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 17, 937–946 (2021).
Zebisch, M. et al. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 4, 2787 (2013).
Marusyk, A., Janiszewska, M. & Polyak, K. Intratumor heterogeneity: the Rosetta stone of therapy resistance. Cancer Cell 37, 471–484 (2020).
Levengood, M. R. et al. Orthogonal cysteine protection enables homogeneous multi-drug antibody-drug conjugates. Angew. Chem. Int. Ed. Engl. 56, 733–737 (2017).
O’Brien, C. et al. Functional genomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res. 68, 5380–5389 (2008).
Chen, R. et al. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol. Cancer Ther. 14, 1376–1384 (2015).
Okeley, N. M. et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 16, 888–897 (2010).
Li, F. et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 76, 2710–2719 (2016).
Doronina, S. O. et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 17, 114–124 (2006).
Walker, J. A. et al. Substrate design enables heterobifunctional, dual “click” antibody modification via microbial transglutaminase. Bioconjug. Chem. 30, 2452–2457 (2019).
Sabbaghi, M. et al. Defective cyclin B1 induction in trastuzumab-emtansine (T-DM1) acquired resistance in HER2-positive breast cancer. Clin. Cancer Res. 23, 7006–7019 (2017).
Cilliers, C., Menezes, B., Nessler, I., Linderman, J. & Thurber, G. M. Improved tumor penetration and single-cell targeting of antibody-drug conjugates increases anticancer efficacy and host survival. Cancer Res. 78, 758–768 (2018).
Yuan, R. et al. 2022 – Abstract and Poster Presentation: Th42 – Next-generation immunostimulatory antibody-drug conjugate (iADC) combines direct tumor killing and innate immune stimulation to provide protective anti-tumor immunity (2022).
Kumar, A. et al. Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg. Med. Chem. Lett. 28, 3617–3621 (2018).
Nilchan, N. et al. Dual-mechanistic antibody-drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib. Ther. 2, 71–78 (2019).
Tang, C. et al. One-pot assembly of dual-site-specific antibody–drug conjugates via glycan remodeling and affinity-directed traceless conjugation. Bioconjug. Chem. 34, 748–755 (2023).
Acknowledgements
The authors acknowledge support from the US NIH (R35GM138264 and R01CA283876 to K.T.) and the US Department of Defense Breast Cancer Research Program (W81XWH-19-1-0598 to K.T.).
Author information
Authors and Affiliations
Contributions
All authors made a substantial contribution to all aspects of the preparation of this manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors are named inventors on all or some of the patent applications (WO2018218004A1, US11629122B2, EP3630189A4 and WO2023122587A3) relating to the linker technologies described in this article. K.T. is a co-founder of and holds equity in CrossBridge Bio.
Peer review
Peer review information
Nature Reviews Clinical Oncology thanks N. Tumey and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tsuchikama, K., Anami, Y., Ha, S.Y.Y. et al. Exploring the next generation of antibody–drug conjugates. Nat Rev Clin Oncol 21, 203–223 (2024). https://doi.org/10.1038/s41571-023-00850-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41571-023-00850-2
This article is cited by
-
Cancer therapy with antibodies
Nature Reviews Cancer (2024)
-
How antibody–drug conjugates aim to take down cancer
Nature (2024)
-
Design, synthesis and bioactivity evaluation of novel monomethyl auristatin F analogues
Molecular Diversity (2024)
