
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused millions of deaths and substantial morbidity worldwide. Intense scientific effort to understand the biology of SARS-CoV-2 has resulted in daunting numbers of genomic sequences. We witnessed evolutionary events that could mostly be inferred indirectly before, such as the emergence of variants with distinct phenotypes, for example transmissibility, severity and immune evasion. This Review explores the mechanisms that generate genetic variation in SARS-CoV-2, underlying the within-host and population-level processes that underpin these events. We examine the selective forces that likely drove the evolution of higher transmissibility and, in some cases, higher severity during the first year of the pandemic and the role of antigenic evolution during the second and third years, together with the implications of immune escape and reinfections, and the increasing evidence for and potential relevance of recombination. In order to understand how major lineages, such as variants of concern (VOCs), are generated, we contrast the evidence for the chronic infection model underlying the emergence of VOCs with the possibility of an animal reservoir playing a role in SARS-CoV-2 evolution, and conclude that the former is more likely. We evaluate uncertainties and outline scenarios for the possible future evolutionary trajectories of SARS-CoV-2.
Similar content being viewed by others

Introduction
The COVID-19 pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a betacoronavirus, which is closely related to the human SARS-CoV virus — the cause of the 2002–2004 SARS outbreak. Three years since the start of the first coronavirus pandemic in living memory, attention understandably turns to what a future with the SARS-CoV-2 virus might look like. The pandemic also saw the generation of unparalleled amounts of genomic data for a single pathogen1, serving to combat but also understand the biology of this virus. We witnessed evolutionary events that have previously been largely the preserve of indirect inference, including the diversification of SARS-CoV-2 into variants with distinct phenotypic characteristics including transmissibility, severity and immune evasion. Tracking the evolution of this pathogen in real time offers hope of understanding the processes generating this diversity, potentially predicting possible future evolutionary trajectories of the virus, and offering avenues for prevention and treatment. To facilitate such possibilities there is a pressing need to critically review the key drivers of SARS-CoV-2 evolution, and to explain the processes that generate diversity and novelty in the virus.
Like most RNA viruses, coronaviruses evolve rapidly, their evolution occurring on timescales of months or years and often observable and measurable. Evolution occurs on comparable timescales with the virus’ transmission events and ecological dynamics (such as changes in the number of infectious individuals over time, immunity profiles and human mobility). As a consequence, evolutionary, ecological and epidemiological processes impact each other, a feature of RNA viruses2. Evolution in viruses is driven by the rate at which mutations are generated and spread through populations. Natural selection will act to fix advantageous mutations, such as, for example, the D614G mutation, which confers elevated transmissibility3. Viral evolution involves an additional level of complexity, as viruses replicate and evolve within individuals, but they must also successfully transmit person to person, resulting in evolution at a different scale. Most variation is lost during the tight bottlenecks imposed at transmission, whereas some mutations are often passed on by chance, without selective advantage4. In addition to these population-level processes, as viral lineages diversify, including into potentially antigenically distinct strains, higher-level processes such as lineage competition and extinction emerge.
In this Review, we consider the evolution of SARS-CoV-2 at different scales, the phases of the COVID-19 pandemic, factors that drive the evolution of the virus, theories for the emergence of epidemiologically important variants and potential future evolutionary scenarios and their likely public health repercussions.
The generation of diversity during the SARS-CoV-2 pandemic
Mutation rate: replication fidelity and host-mediated genome editing
A key determinant of the rate at which a virus evolves is its mutation rate. This is the intrinsic rate at which genetic changes emerge per replication cycle, a biochemical property determined by the replication fidelity of a virus’ polymerase enzyme. These genetic changes are the ‘raw material’ on which selection acts. Most mutations are deleterious, and virions hosting them fail to replicate5,6,7. SARS-CoV-2 mutation rate estimates of around 1 × 10–6–2 × 10–6 mutations per nucleotide per replication cycle are consistent with previous estimates in other betacoronaviruses5,8,9. These mutation rates lie below the range of rates that are typical for other RNA viruses such as hepatitis C virus (HCV; ~10–5 × 10–6 mutations per nucleotide per replication cycle) and human immunodeficiency virus (HIV; ~10–4 × 10–6 mutations per nucleotide per replication cycle), which, unlike coronaviruses, lack a 3′ exonuclease proofreading mechanism in their replication machinery8,10,11,12. Insertions and deletions result from replication errors and can also generate diversity, such as the deletion at position 69–70 of the spike gene responsible for the S-gene drop-out that was instrumental in detecting the SARS-CoV-2 Alpha variant, and has been reported to be associated with increased infectivity13.
In addition to RNA replication errors, host-mediated genome editing by innate cell defence mechanisms may introduce substantial numbers of directed mutations into the SARS-CoV-2 genome, and thus may influence its evolutionary rate. Cellular mutational drivers include members of the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family14,15,16, including APOBEC1, APOBEC3A and APOBEC3G that demonstrate editing activity for numerous DNA and RNA virus and retroviral genomes17,18, including SARS-CoV-2 (ref. 19). APOBEC activity has been inferred bioinformatically through observations of a substantial excess of C → U transitions over all other mutations18,20,21. SARS-CoV-2 genomes may also be edited by different cellular antiviral proteins (adenosine deaminases that act on RNA 1 (ADAR1)), leading to A → G mutations (and U → C mutations in opposite genomic strands)21,22.
The potential editing-associated C → U mutations in the SARS-CoV-2 genome sequences introduce complexities to SARS-CoV-2 evolutionary genomic analysis. C → U mutations account, in part, for the strikingly high ratio of non-synonymous changes in SARS-CoV-2 genomes compared with those at synonymous sites; the mean dN/dS ratio is ~0.7–0.8, which is a measure of the ratio of non-synonymous mutations per non-synonymous site (dN) to synonymous mutations per synonymous site (dS)20. Such mutations may be a potent driver of antigenic or phenotypic changes. Furthermore, C → U mutations may be skewed towards mutational ‘hot spots’ generated by RNA structures and specific base contexts. Repeated cycles of C → U transitions and selective reversions may create a large number of homoplasic sites20,23,24 and, therefore, convergence in otherwise genetically divergent strains.
Substitution rate
Although often confused, the substitution rate (also known as the rate of molecular evolution) is distinct from the mutation rate25. The substitution rate measures the pace of mutation accumulation as the virus evolves. A high rate means the virus accrues many mutations per unit of time. For RNA viruses, the substitution rate is commonly estimated using phylogenetic methods. In essence, these employ statistical phylogenetics, combining information on time spans and differences in the number of mutations in virus sequences sampled at different time points to estimate a substitution rate26,27. Importantly, only mutations that reach detectable frequencies in the population contribute to estimations of the evolutionary rate (Fig. 1). An important obstacle to measuring the SARS-CoV-2 substitution rate during the early stages of the pandemic was the limited amount of accrued evolutionary changes, insufficient to make robust estimations28. Before the emergence of variants of concern (VOCs), the virus was estimated to acquire nearly two evolutionary changes a month (~2 × 10–6 per site per day)28,29 (Fig. 1a).

a, As the virus spreads and acquires further evolutionary changes, sequence data from different time points can be used to infer viral evolutionary rates as well as the time to the most recent common ancestor (TMRCA) of sampled viruses. The rate of evolution is elevated at terminal branches (shown in red) relative to internal branches (shown in blue). This is because the former includes more deleterious mutations that only persist in the population for a limited period of time before they are removed by purifying selection. b, If the genomic samples include both variant of concern (VOC) and non-VOC (background) sequences, the evolutionary rate at the stem branch (shown in red) connecting the clade of the VOC sequences to the rest of the tree is elevated in comparison with the rate on all other branches in the entire phylogeny. This is because of the large number of evolutionary changes present in all VOC samples relative to the background.
Recombination
Recombination is another mechanism that can expedite adaptation in viruses by bringing together mutations from different genetic backgrounds to create hybrid variants. Recombination is a common feature of betacoronavirus evolution and has been detected in SARS-CoV-2 (ref. 30) and other sarbecoviruses30,31,32. In order for recombination to occur and be subsequently detected, a host must be co-infected with two genetically distinct viruses that, when recombined, produce viable progeny that can spread to other hosts. Our ability to detect recombinants therefore increases over time since emergence, along with a growing genetic divergence of SARS-CoV-2, which allows multiple divergent lineages to co-circulate within the same region (Fig. 2d).

a, At the start of an acute infection, viral load increases rapidly for a period of time (tg) before the immune system begins to clear the infection, at which point the viral load starts to decrease until it returns to levels to below the detection threshold over a period of time (tc). During these periods, new variants may emerge as a result of mutations (virions in colour). b, At the point of transmission, major variants that are present at high frequency in the infection donor are more likely to pass through the narrow transmission bottleneck. However, sometimes a minor variant is passed on to a recipient host (shown in red) resulting in mutations getting passed on to the rest of the population. This illustrates the role of chance introduced by the transmission bottleneck in the evolution at the host population level. c, As the number of new infections grows over the course of an epidemic wave, new variants with selective advantage may emerge and reach high frequencies. Others, carrying deleterious mutations, get purged from the population. Superspreading events and transmission bottlenecks introduce an element of chance in determining the frequencies of variants in the population over time. d, As more variants emerge in the population, they can go on to seed new outbreaks in different geographical regions, which may lead to two or more variants co-circulating in the same region. Their genetic differences allow us to identify these different variants, infer their evolutionary origin and track their spread. When two or more distinct variants co-circulate in an area, this increases the risk of emergence of novel variants as a result of recombination.
One of the first reported cases of inter-lineage SARS-CoV-2 recombinants was the XA lineage, first detected in the United Kingdom33,34. Tentative evidence of recombination between VOCs (Alpha and Delta) has also been reported in a small cluster of cases in Japan35. A later study showed widespread circulation of a recombinant B.1.631/B.1.634 lineage (designated as lineage XB by the Pango nomenclature) in North America. Later studies found three recombinant lineages, which were given Pango designation. Two are a combination of Delta and BA.1 (XD and XF) and one is a BA.1/BA.2 recombinant (XE)36. More recently, several other Omicron recombinants have been identified37.
Levels of evolution in SARS-CoV-2
Evolution within individuals during acute infections
The majority of infections with SARS-CoV-2 are acute and cleared by the immune system typically within 10–15 days after the onset of symptoms38,39,40 (Fig. 2a). Once SARS-CoV-2 infects an individual, viral particles are produced exponentially in the respiratory tract, reaching peak titres around 2–5 days post infection, which approximately coincides with the time of symptom onset41,42, with similar dynamics across most variants of SARS-CoV-2 (ref. 43), except for Omicron that peaks around 3 days after the onset of symptoms44.
Within-host diversity of viruses is usually quantified by the number of intra-host single-nucleotide variants (iSNVs) that are detected above a certain minor allele frequency threshold (usually >2–5%). During a typical acute infection, SARS-CoV-2 intra-host diversity is limited, with most samples containing very few iSNVs at low frequency45. There is also likely tissue organ compartmentalization of the virus, as demonstrated by discordance between the viral populations in nasal and oral environments46,47.
Transmission bottleneck
The transmission bottleneck is the amount of genetic diversity in the founder virus population that gets transmitted to a new host, compared with that in the donor host in a transmission event. The transmission bottleneck is therefore the link between within-host evolutionary processes and the between-host level of evolution. Following transmission, SARS-CoV-2 infection is typically established by one or two virions, meaning most variants generated during the course of the previous infection are lost, or occasionally fixed in the new host if they happen to be transmitted. This implies that iSNVs are rarely shared between individuals45,48 (Fig. 2b). The considerable role of stochasticity in the transmission of iSNVs through the transmission bottleneck impedes robust estimations of the magnitude of the selective advantage of mutants, except when selection is very strong49,50. D614G, and to a greater extent the emergence of VOCs, all featured striking selective advantages. A narrow transmission bottleneck is a universal feature of viral transmission51, observed in human52,53,54,55 and non-human viruses alike51,56.
Evolution at the host population level
When considering evolution at the between-host scale, within-host viral diversity is typically ignored and, instead, focus is given to the consensus sequence, which is essentially the sequence obtained by taking the most common iSNV at each site along the genome. Before the emergence of VOCs, and now within the major VOC lineages, the limited genetic diversity of the virus present in individuals who are acutely infected and the narrow transmission bottleneck mean that most of the observed genetic diversity at the between-host consensus level represents neutral or slightly deleterious mutations that overcame the narrow transmission bottleneck owing to chance. This stochasticity enables mutations without a strong selective advantage to circulate in the population and also reach high frequencies by chance, a process known as genetic drift. As well as the narrow transmission bottleneck, ‘superspreading’, whereby a small fraction of infectious hosts are responsible for the majority of transmissions57,58, is a further source of stochasticity and, hence, also contributes to genetic drift. Superspreading events increase stochasticity by introducing heterogeneity in the number of secondary infections, which in turn reduces the effective population size of a virus59.
The narrow transmission bottleneck often produces a founder effect, as only one or a few ‘founder’ viruses are the ancestors of all viruses during the new infection, and all infections in the subsequent chain of transmission. If a new outbreak is ultimately caused by a single founder-source individual, then all subsequent infections will have a similar viral consensus genotype. During the early stages of the pandemic, it proved difficult to establish whether a variant was increasing in frequency because it had an intrinsic advantage or due to factors such as drift or founder effects. In particular, the global fixation of the D614G mutation in early 2020 sparked debates about whether it was a result of natural selection or chance60. Later studies showed that this mutation actually gave this variant a near 20% transmissibility advantage over the original B.1 lineage61,62 (Fig. 2c,d).
Evolutionary phases of the pandemic
A period of apparent evolutionary stasis
After the emergence of SARS-CoV-2 in humans, for the first nearly 8 months the virus seemed to exhibit limited apparent evolution. This was partially due to the relatively small global virus population, while spread was still not ubiquitous, and later as a result of non-pharmaceutical interventions in many parts of the world, and partially an artefact of virus undersampling. These factors, along with prior knowledge of the proofreading capacity of the coronavirus polymerase enzyme, led at the time to expectations that SARS-CoV-2 will evolve slowly, and that evolution will not play an important role in the unfolding and control of the pandemic. With the D614G substitution being the most noticeable evolutionary change in April 2020, this period was characterized by limited examples of viral diversity and evolution.
Over this time period, the estimated substitution rate of SARS-CoV-2 decreased by nearly 50%. This was mainly the result of incomplete purifying selection29, which over short timescales leaves an overabundance of not yet purged deleterious mutations in the virus population. For this reason, the rates of evolution at smaller timescales, represented by the terminal branches on a phylogenetic tree, are elevated relative to longer-term evolution, represented by the internal branches in the phylogeny (Fig. 1a). This phenomenon is likely responsible for altering the estimated substitution rate also of other viruses over the course of epidemic waves29,63,64.
Emergence of highly divergent lineages
It took 8 months for the first divergent SARS-CoV-2 lineages to appear (Fig. 3a), marking a turning point in the pandemic from an evolutionary point of view. The first three such lineages, later termed VOCs Alpha, Beta and Gamma, emerged independently in different parts of the world and were the result of puzzling higher evolutionary rates. The sheer number of mutations involved in VOCs is particularly striking from an evolutionary point of view. Alpha and Gamma feature, respectively, 14 and 11 extra non-synonymous mutations relative to their ancestral lineages whereas Omicron had more than a few dozen extra mutations in the spike gene65. These observations appear to have been generated by unusual circumstances most consistent with continued replication during chronic infections allowing the virus to acquire many evolutionary changes. This contrasts with the chain of acute infections typical for a respiratory virus, which enforces tight bottlenecks at transmissions, periodically purging mutations (see section on ‘Evolutionary origins of variants of concern’).

a, Frequency of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) lineages over time. b,c, Accumulation of amino acid mutations in the S1 subunit over time (panel b) and as a function of their relative mutational fitness (panel c) shown for 3,055 globally representative genome samples of SARS-CoV-2 between December 2019 and December 2022 generated by nextstrain.org182. Mutational fitness is calculated using results from ref. 105 that computes the relative impact of each mutation in the growth advantage of a lineage using a hierarchical Bayesian regression model. The model estimates the exponential growth advantage of each SARS-CoV-2 lineage (proxy for measuring relative fitness) as a linear combination of the effect of individual mutations. Each Pango lineage (World Health Organization (WHO) label, Nextstrain clade) is given a unique colour. VOC, variant of concern.
Although there is no significant difference between the overall evolutionary rate estimates for the non-VOC background and inside the VOC clades of the SARS-CoV-2 phylogenetic tree33,66,67, the substitution rate of the stem branch connecting the background to the VOC clades is approximately twofold to fourfold higher67,68 (Fig. 1b). This difference in evolutionary rate is only seen for non-synonymous substitutions, whereas the rate of synonymous substitutions is largely similar relative to within VOC clades and the non-VOC clades69.
Gradual within-lineage evolution
The discovery of Omicron in late November 2021, originally comprising three sister lineages (BA.1, BA.2 and BA.3), marked the start of a new phase of the pandemic, which, dissimilar to the preceding one that gave rise to highly divergent lineages, was dominated by successive sweeps of Omicron sub-lineages. Soon after BA.1 reached global dominance, it was replaced by BA.2, which further diversified into sub-lineages including BA.2.12.1 and BA.2.75, and by BA.5, which reached high prevalence globally and is phylogenetically distinct from BA.2 sub-lineages70. Since the global dominance of BA.5, several sub-lineages of Omicron have emerged, but none of them have yet successfully outcompeted BA.5. Instead, they exhibit remarkable convergent evolution, with multiple shared mutations in the spike gene71.
Throughout 2021 and 2022, the evolution of SARS-CoV-2 was characterized by a steady increase in divergence within major lineages and a stepwise increase associated with each successive new major lineage, leading to a faster overall rate of evolution. These between-lineage evolutionary dynamics are compatible with a molecular clock that is substantially faster than the within-lineage rate66,69. However, after the emergence of BA.5, it is now unclear whether SARS-CoV-2 will continue to evolve in this saltatory fashion with repeated emergence of highly divergent lineages, or whether it is transitioning to a more gradual adaptive process. In 2022, multiple lineages emerging within BA.2 and BA.5 were observed, in a more stepwise fashion, with several amino acid changes and moderate transmission advantages, which could indicate a shift to a more gradual stepwise evolution (see ‘Possible future scenarios’ for more discussion on this).
Transmissibility: the primary driver of SARS-CoV-2 evolution
Evolution of intrinsic transmissibility
Parasites typically exist in a population of hosts, a highly fragmented environment with discrete and ephemeral habitats, where their intrinsic ability to transmit is a crucial fitness element72, particularly for obligate parasites such as viruses. For viruses causing acute infections, where the period of communicability is short, high transmissibility is an overriding trait73. In these viruses, transmissibility — usually expressed as the net reproduction number (Rt; the total number of secondary infections each case generates in a population) — is assumed to closely approximate their fitness at the host population level74. The continuous evolution of these kinds of viruses towards higher transmissibility therefore can be understood as a straightforward evolutionary process of fitness maximization.
The process of transmission can be divided into three steps: shedding of the virus from the infectious host; its survival and travel in the environment; and its establishment in the recipient macroorganism. Natural selection operates on specific traits of the virus that can facilitate each of these steps and increases in intrinsic transmissibility are the result of ongoing evolution in these transmission-enhancing traits (Fig. 4).

a, Relationships between the key elements that contribute to transmissibility: intrinsic transmissibility, immune escape and temporal aspects of infectiousness, such as time of onset and duration of infectiousness. b, Evolvable viral traits affect intrinsic transmissibility of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by influencing mechanistic factors contributing to the transmission process at each of its three stages. Evolution in these SARS-CoV-2 traits can ultimately increase transmissibility. c, Transmission-enhancing properties of SARS-CoV-2 influenced by viral evolution, and their effect on important epidemiological parameters and population outcomes. aOnly in combination with immune escape. ACE2, angiotensin-converting enzyme 2; R0, basic reproduction number; Rt, net reproduction number.
Optimizations of one such trait, for example the interaction between SARS-CoV-2 and the angiotensin-converting enzyme 2 (ACE2) receptor (its primary cell entry route), increases the transmissibility of the virus in two ways: it elevates infectiousness by increasing the number of infected cells, thus boosting viral loads in mucosal secretions of infectious individuals; and it also enhances the ability of the viral lineage to establish infection in the new host75,76,77 (Fig. 4b). Mutations in the spike protein can enhance and stabilize its binding to the receptor. This was first observed with mutation D614G78,79. Later, VOCs Alpha, Delta and Omicron were found to carry mutations that further improved binding, N501Y in the receptor binding domain (RBD) being the best example80.
An essential factor for cell entry, through mediating membrane fusion, is the cleavage of the spike protein81. In the ferret model, the furin cleavage site insertion was essential for virus transmission82. Mutations P681H in Alpha and Omicron, and P681R in Delta, render the spike protein nearly fully cleaved, thus facilitating viral entry and, ultimately, intrinsic transmissibility83. Overall, mutations promoting receptor binding and spike cleavage seem to increase both infectiousness and infectivity, and improve the spread of the respective lineages. Nucleocapsid mutations (R203K + G204R) could enhance replication and transmissibility84. In Alpha, evolution outside spike seems to increase subgenomic RNA levels for the nucleocapsid, ORF9b and ORF6 genes, leading to innate immune escape and improved transmission85.
Viruses can also enhance transmissibility by evolving tropism for a tissue or organ, which may be a better disseminating platform. Unlike the ancestral SARS-CoV-2 virus, which infected bronchial and lung cells, Omicron BA.1 evolved a preference for efficient replication in the nasopharynx, a better vantage point for entering aerosol86. Omicron BA.1 also appears to replicate faster than other VOCs in ex vivo bronchi cultures, but poorly in lung cells87.
Virion stability outside the host is also an integral component of intrinsic transmissibility and likely impacts viral fitness considerably. The ability of SARS-CoV-2 to remain in aerosols has been demonstrated to differ across lineages in the pandemic88. Studies of aerosol stability reveal a longer half-life presence of Alpha and Beta in comparison with the ancestral lineage, whereas Delta and Omicron were of comparable stability with it89. Another study found extremely low and similar virion longevity in aerosols across VOCs, further suggesting that unless differences between evolving lineages are considerable, stability in aerosols may not be a decisive factor in the evolution of transmissibility90.
Other than being intrinsically more transmissible, a virus can maximize its reproduction number (Rt) via prolonged infectiousness. The longer a host is infectious, the more secondary infections it can cause, thus increasing its Rt. Duration of infectiousness is therefore itself an evolvable trait91.
Considering two viruses with the same levels of intrinsic transmissibility, one may transmit faster than the other, provided its period of infectiousness starts earlier. The latent period is an epidemiological property, representing the time between the moment of infection of an individual and the moment they start to be infectious to others. A shorter latent period means the host can infect sooner after being infected92, and for a given level of Rt this can cause epidemics with steeper growth93. An earlier onset of infectiousness was observed for Omicron BA.1 when compared with Delta94, yet its infectiousness was shown to last a shorter time95.
Transmissibility in immune populations: evolution of immune escape
RNA viruses are known to exhibit considerable degrees of antigenic evolution — adaptive changes in genomic regions encoding for targets of immunity. Antigenic drift often results in immune escape — the failure of humoral or cellular immunity to recognize or neutralize the pathogen. Whereas in naive host populations intrinsic transmissibility is the dominant adaptive property of viruses, in highly immune populations the ability to overcome host resistance becomes at least equally important as a fitness determinant. Even a highly transmissible virus will not be able to spread among resistant hosts. Allowing reinfection of immune individuals, immune escape mutations are effectively opening a new ecological niche for escape lineages — the niche of reinfections.
In SARS-CoV-2, signs of antigenic evolution were identified in late 2020 in VOCs Beta and Gamma, each found to carry mutations demonstrated to reduce antibody recognition and neutralization, particularly the E484K mutation. Early incidence data from South Africa and Brazil — the respective areas where these lineages were first identified — indicated higher reinfection rates in comparison with areas where other lineages circulated, illustrating the important role of immune escape mutations in maintaining high transmissibility in immune populations (Fig. 4c).
On emergence of Omicron in autumn 2021, it was quickly realized that this variant has a much higher capacity to cause reinfections than any variant before it96. Many of the major VOC mutations in the spike are found in the RBD and amino-terminal domain where neutralization antibody binding is the most potent97. Deep mutational scanning studies have provided rich data on the ability of these mutations to increase ACE2 binding affinity98, and to escape antibody binding99. In particular, they show the major impact of E484 (amino acid changes to K, P and Q) and N501 (amino acid changes to Y and T) sites on plasma antibody neutralization and ACE2 binding affinity of the virus, respectively.
With more than 30 amino acid substitutions and several deletions and insertions, the first 2 lineages of Omicron (BA.1 and BA.2) were significantly more divergent in relation to earlier VOCs65. The new mutations that arose in these Omicron sub-lineages, also mainly clustering in the RBD, have caused significant reductions in the neutralization titres of sera from individuals who are naturally infected or vaccinated100,101 but may have only had marginal influence on their ACE2 binding affinity102. The descendants of BA.4 and BA.5 lineages contain further mutations in the RBD relative to the earlier Omicron lineages including L452R and F486V, shown to contribute significantly to their immune escape properties103. Sera from individuals who are vaccinated and boosted also exhibited reduced neutralization of these lineages, compared with BA.1 and BA.2 (ref. 103).
The S1 subunit, containing the RBD and N-terminal domain that possess a substantial number of mutations shared among these new highly divergent and evasive variants, exhibits a strong signal of adaptive evolution, mainly reflecting the increased ability of these lineages to transmit in immune populations, and particularly after the emergence of Omicron104 (Fig. 3a). Predictably, these mutations were also associated with aspects of increased fitness105 (Fig. 3c).
The contribution of viral escape from cell-mediated immunity in driving the evolution of SARS-CoV-2 is less well defined than that of escape from humoral immunity. The majority of T cell epitopes are invariant between the prototype strains and VOCs106,107,108. The spike mutation P272L was shown to result in immune escape of a dominant T cell epitope109, and several other mutations in T cell epitopes reduced or directly abrogated MHC class I presentation110.
Besides natural immunity, vaccination can also be a driver for immune escape. The crucial difference for SARS-CoV-2, in particular, is the lack of mucosal immunity following parenteral vaccination. Because the virus can still replicate in the mucosa of the upper respiratory tract and transmit, the role of vaccination as a factor driving SARS-CoV-2 evolution may be less pronounced compared with that of natural infection. Another important difference to natural infection is the much narrower antigenic region targeted by most popular vaccines, that is, the spike protein or even just the RBD. This naturally limits the drive for escape pressure to just these regions. Vaccination-related immune escape has nevertheless been shown for both Beta and Omicron111, expectedly with a focus on the anti-RBD-induced antibodies. Escape from vaccine-elicited humoral immunity was further demonstrated for the Delta variant112. While mass vaccination with ancestral strains may create a more constant and predictable immune pressure, the spread and ongoing evolution of the pathogen render natural immunity a much more dynamic selective force. The changing immune landscape will mean that, at any time, a variant that has high escape to its immune landscape will spread rapidly through the population and can, potentially, outcompete variants triggering immunity.
Waning immunity, which is the decline of immune protection over time and characteristic for immunity to SARS-CoV-2, is a factor likely to slow down the population-level evolution of immune escape of the virus. Because of waning immunity, those fully resistant to reinfection will typically be fewer than those with partial immunity, so intrinsically transmissible viral lineages can maintain high fitness even without immune escape in non-naive populations.
The evolution of virulence
The term virulence is defined differently across disciplines. In ecology, virulence is formally defined as the degree of reduction in the fitness of a host attributed to a parasite113. In clinical medicine and experimental health sciences, often the synonym ‘pathogenicity’ is preferred to denote the degree of harm a pathogen causes to a host. Pathogenicity in clinical medicine can also be described in terms of the specific symptoms a pathogen causes. Unlike virulence and pathogenicity, which characterize the pathogen, the related term ‘severity’ is used to describe the gravity of a clinical condition.
Virulence is not an actual trait in the biological sense. It is, rather, an interaction property, that is, the product of the ecological relationship between two species — a host and a parasite. This ‘relativity’ of virulence is well illustrated by the fact that the same pathogen can exhibit very different levels of virulence when infecting different host species. As an ecological outcome from complex, multifactorial interactions, virulence is difficult to model or predict, but an understanding of its component processes can, in part, provide some predictions about its evolution.
Changes in pathogenicity were first reported for Alpha. Thereafter, all subsequent VOCs before Omicron (Beta, Gamma and Delta) were causing increased hospitalizations and mortality rate in comparison with the ancestral lineage114,115,116. The later emerging Omicron BA.1 and BA.2 lineages were both associated with lower disease severity compared with the ancestral strain36,117, and virulence levels of different variants did not exhibit any directional pattern. Such comparisons are of course challenging as not only the viral lineages are subject to change. So is the resistance status of hosts, due to widespread vaccination or previous infections, meaning that Omicron spread has occurred against a background of much higher population immunity levels. Most virulence studies for SARS-CoV-2 are understandably focused on the role of the spike protein, but other parts of the genome also contribute to this property. In animal models, chimeric viruses carrying the spike protein of Omicron BA.1 within a backbone genome from the ancestral virus demonstrated the existence of virulence factors in other parts of the SARS-CoV-2 genome in addition to the important role of the spike protein for virulence118,119,120.
A popular and incorrect view on the evolution of virulence, frequently expressed in the context of SARS-CoV-2, is that in the long run, pathogens will tend to evolve to be decreasingly virulent121. The reasoning being that highly virulent pathogens will short-sightedly kill their host and inevitably perish with it. There are crucial flaws in this oversimplistic logic. First, it ignores the fact that the actual adaptive environment of viruses is not a single host but a population of hosts. For many pathogens, severe disease manifestations postdate transmission to a new host. SARS-CoV-2 tends to cause severe disease or death late, towards the third week post infection, whereas the infectious period usually spans from day 2 to day 15, with 90% of transmission already achieved before the average time of death. As long as a viral lineage successfully carries on transmitting further to multiple other hosts, the ultimate fate of the initial host will not substantially impact its fitness. In this situation, high virulence is not a fitness impediment for the virus and would not be selected against.
Except in rare and unusual circumstances122,123, microorganisms do not directly benefit from virulence. Yet it could correlate with other traits of the pathogen, which are adaptive. In other words, increasing virulence can be a by-product of viral evolution, where the virus evolves to maximize other traits that increase its fitness, but that are linked to virulence. An example of this situation is SARS-CoV-2 viral loads. Increased viral abundance contributes to better chances of transmission — a crucial fitness trait for the virus. Yet higher loads may also result in more severe disease. In such a situation, a virus may evolve higher virulence, if there is a net gain in fitness.
An additional important and underappreciated point related to virulence is that highly transmissible pathogens (whether due to high intrinsic transmissibility or immune escape) with lower infection fatality ratios can contribute to high population-level disease burdens, overshadowing in that respect extremely pathogenic but less transmissible pathogens (Fig. 4c). Examples of this are MERS-CoV and SARS-CoV, where the former, despite a staggering infection fatality ratio of more than 30% has, due to its relatively low transmissibility, caused a total of 935 deaths since 2012 (ref. 124). By contrast, SARS-CoV-2, with an estimated infection fatality rate well under 1%, has as of today killed more than 18 million people125.
Due to the multifactorial and ecological nature of virulence, and due to the paucity of reliable estimates for many of the parameters involved, the evolution of virulence is difficult to model or predict. Given the relative timing between transmission and severe disease, and life history links between virulence and adaptive traits, we know we cannot rely on evolutionary forces to necessarily reduce virulence as the virus adapts long term to its host population. Depending on a combination of specific circumstances, SARS-CoV-2 virulence could go up or down.
Evolutionary origins of variants of concern
From Alpha to Omicron
In late December 2020, a new SARS-CoV-2 lineage was identified to be expanding rapidly in parts of the United Kingdom, carrying a large number of mutations in the spike region126,127. This lineage, later Pango classified as B.1.1.7 (ref. 128), was named by the World Health Organization (WHO) VOC Alpha129. In the ensuing weeks, South Africa and Brazil reported two additional rapidly growing lineages — VOCs Beta (Pango lineage B.1.351)114 and Gamma (Pango lineage P.1)130. Each of these featured a large number of genetic differences with respect to the background viral population, some bearing signatures of enhanced transmissibility or immune escape properties131,132. The Delta lineage (Pango lineage B.1.617.2), recognized as a VOC in May 2021 but circulating for months before this in India133, rapidly replaced previous VOCs and led to a drastic surge in cases around the world134,135. In November 2021, Omicron65,136 (Pango lineages BA.1–BA.5), discovered in South Africa and Botswana, started new global waves of infection. Although these VOCs emerged in different parts of the world, they shared sets of mutations (for example, N501Y, E484K and ΔH69/V70), indicating possible convergent evolution97,137,138. Each of these VOCs had a significantly higher growth advantage relative to their predecessor variants105.
Although Alpha was the first VOC to be discovered, phylogenetic estimates suggest Beta likely emerged earlier, before June 2020 (ref. 114), months before it was reported in October 2020. The emergence of Alpha was estimated to be in early September 2020 and Gamma in mid-November 2020 (refs. 68,139). Intermediate Alpha-like and Gamma-like genomes were detected, appearing several months before their respective VOC clades first emerged68,139. The origin and beginning of the spread of Delta within India are uncertain, but phylogenetic estimates based on global data suggest that it emerged in mid-October 2020 (ref. 140). Unlike the other VOCs, following its discovery Delta did not increase substantially in frequency until much later in March 2021 (ref. 140). The first three lineages of Omicron (BA.1, BA.2 and BA.3) all emerged independently around the same time in October 2021 (ref. 65), followed by BA.4 in mid-December 2021 and BA.5 in January 2022 (ref. 136). The emergence of the BA.3 lineage has been suggested to be a result of an ancestral recombination event between BA.1 and BA.2 (ref. 65). Also, the emergence of the newly identified BA.4/BA.5 lineages was likely through a prior inter-lineage recombination event114.
The mechanisms of the evolutionary origin of VOCs are still a matter of debate. Several hypotheses have been put forward to explain their emergence: sustained stealth circulation of SARS-CoV-2 in humans in areas with poor genomic surveillance; zoonotic circulation of SARS-CoV-2 in animal reservoirs; and chronic SARS-CoV-2 infections in certain individuals who are immunocompromised (Fig. 5).

Hypothesis 1: the virus can circulate, causing acute infections within a region lacking adequate genomic surveillance for a sustained period of time and gradually acquiring adaptive changes without being detected until it reaches another region with stronger genomic surveillance. Hypothesis 2: people in close contact with farmed or wild animals such as mink or deer can transmit the virus to these populations, where severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can acquire further adaptive changes on circulation in those animal populations before a spillback into humans, leading to the emergence of variants of concern (VOCs). Hypothesis 3: if the virus persists in an individual who is chronically infected for a long period, it may acquire changes that are adaptations to host immune responses. Further adaptive changes may occur if the patient is under treatment to clear the infection (for example, monoclonal antibodies or convalescent plasma) that may accelerate selection for antibody evasion mutations.
Hypothesis 1: undetected circulation in humans
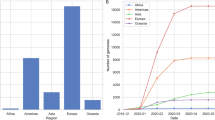
The global genomic surveillance of SARS-CoV-2 has been overwhelmingly more detailed than that of any other pathogen (Fig. 3), yet extremely uneven with many low- and middle-income countries sequencing <0.5% of their cases. This extreme undersampling leads to some viral lineages circulating undetected, allowing for long-term stealth viral evolution141,142. This applies particularly for countries with limited genomic surveillance that experienced sustained circulation of the virus during the pandemic143,144. Inadequate genomic surveillance can further under-detect SARS-CoV-2 chronic infections, which can in turn further contribute to undetected viral evolution (see ‘Hypothesis 3: human chronic infections’; Fig. 5).
From an evolutionary perspective, the emergence of a novel and highly transmissible variant with, say, 12 novel mutations through the gradual accumulation of substitutions at a rate of 2 changes per month would require that variant to remain undetected for nearly 6 months before it would be reported. The accrual of mutations could happen faster if some of the mutations are advantageous for the virus and can reach high frequencies faster. However, Delta and Omicron spread throughout the world in a matter of only a few months. Given global interconnectedness, such an evolving lineage is likely to be intercepted at earlier stages of this mutation accumulation process. Furthermore, unless the evolution of the virus is accelerated, a lineage could not acquire 10–12 mutations above what would be expected given the substitution rate just through gradual accumulation of substitutions. Therefore, the emergence of a novel variant in transmission chains of multiple acute infections does not seem likely145.
Hypothesis 2: circulation in animals
Despite a broad range of animal hosts that are permissive to the virus, just three animal species are known to effectively transmit SARS-CoV-2: Syrian hamsters, mink and white-tailed deer — the only known wildlife reservoir at present146,147,148,149. To date, no specific viral genetic adaptations have been observed in the Syrian hamster. Mutations in SARS-CoV-2 found in mink and white-tailed deer both appear animal host-specific. Mutations in isolates from mink improve viral binding to the mink ACE2 receptor, whereas isolates from white-tailed deer are found to carry changes predominantly outside the spike protein150,151. Although the N501Y spike mutation found in VOCs Alpha, Beta and Delta allows these variants to infect wild-type mice, this is likely a coincidence resulting from evolution in human hosts, rather than an adaptation to this animal80. N501Y is also present in the Omicron spike protein, but infection of Balb/C laboratory mice was inefficient with Omicron BA.1. Yet infectivity markedly improved when mice were challenged with a chimeric virus with an Omicron spike and an ancestral backbone120, suggesting that other mutations outside the spike region might also be responsible for susceptibility in mice, a finding that has been confirmed in K18-hACE2 transgenic mice, which express human ACE2 (ref. 119).
Divergent SARS-CoV-2 sequences in farmed mink in the Netherlands and white-tailed deer in Canada show signatures of accelerated evolution and potential for animal-to-human transmission151,152. However, the combination of mutations in these viruses is very different from those found in VOCs in humans, suggesting a different evolutionary path. This is further substantiated by data showing an ongoing adaptation of these strains to a new animal host such as the white- tailed deer151, even if the potential for viral spillback to humans remains.
All pre-Omicron lineages featured similar infection and virulence patterns across Syrian hamsters, K18-hACE2 transgenic mice and ferrets. Yet Omicron BA.1 was unable to infect ferrets, a further indication that its evolution may not be an adaptation to animals118,153,154. Furthermore, passaging of SARS-CoV-2 in, or adaptation to, an animal species is unlikely to elicit human immune escape properties, which is a central feature of most VOCs155. In fact, the numerous mutations that occur predominantly in the SARS-CoV-2 spike protein, driving considerable immune escape in humans, strongly point to long-term evolution in humans, already described in individuals who are immunosuppressed156. Omicron specifically appears to be a product of adaptation to humans, and only few particularly susceptible animal species such as the Syrian hamster can be efficiently infected in the laboratory. Currently, no convincing arguments that support the origin of VOCs in animal reservoirs exist. Nevertheless, due to the longer-term risks of reverse spillover, the evolution of SARS-CoV-2 in new animal reservoirs such as the white-tailed deer needs to be monitored and studied closely.
Hypothesis 3: human chronic infections
Prolonged shedding of SARS-CoV-2 has been reported in individuals who are immunocompromised, due to, for example, some cancers, immunosuppressive therapy or AIDS157,158. In such individuals a deficient immune system can fail to clear the virus on acute infection, leading to long-term viral persistence. Given the large number of amino acid changes in viral lineages sampled from chronic infections, it has been hypothesized that such infections are responsible for the emergence of the multiple highly divergent variants of SARS-CoV-2 (ref. 159). Key evidence in support of this hypothesis is the observation that sets of mutations identified from chronic infections are also shared by VOCs. Examples of such mutations are E484K (seen in Beta and Gamma), N501Y (Alpha, Beta and Gamma), ΔH69–V70 (Alpha, Eta and some Omicron variants), H655Y (Mu) and R346I (Omicron)160. Further, the extremely low number of synonymous mutations accumulated in the spike of Omicron, in stark contrast to the very high numbers of non-synonymous ones69, is consistent with the currently most credible hypothesis for the origin of VOCs, that is, through evolution during chronic infection. A low number of synonymous mutations are a sign of low rates of neutral evolution, which might be an indirect indication of low numbers of transmission bottlenecks in the near past, supporting a scenario of long-term evolution in a persistent infection in a single individual, rather than a chain of transmissions145. Divergent cryptic viral lineages containing previously unsampled mutations isolated from urban wastewater, which come from specific geographically restricted parts of the sewage system, are another indirect signal for possible human chronic infection161.
It is thought that selection in individuals who are chronically infected is likely driven by treatments (Box 1), such as convalescent plasma or monoclonal antibodies, and/or by weak immune responses sufficient to exert a selection pressure on the viral population, but insufficient to clear it, yet other selective pressures such as receptor binding158 or replication capacity are also possible. Given that antibody-based therapies for individuals who are chronically infected predominantly target the SARS-CoV-2 spike protein, we may expect them to select for changes that are concentrated in the spike and for immune escape mutations that will facilitate reinfections, such as those found in many of the VOCs157. Importantly, forward transmission of mutations from patients wo are chronically infected has been detected160,162. Settings with high levels of monoclonal antibody use (perhaps care homes) could have provided an environment that generates viral diversity that can be quickly passed on between individuals.
Although clear evidence of selection has been observed in some individuals who are chronically infected, there is still no consistent understanding of a clear pattern of the evolution of SARS-CoV-2 across all chronic infections, or how common the emergence of transmission-enhancing variants during these infections is. Some of the globally occurring spike mutations in VOCs such as P681H/R, for example, are not observed in some patients with chronic infections. A potential explanation for this may be a trade-off between immune escape mutations and transmissibility, which, unlike viruses in chronic infections, the globally transmitting viral lineages are likely subject to163. It is also unclear which forms of immunosuppression are associated with chronic infection, and their prevalence within the population is uncertain164. Overall, given the current evidence in support of chronic infections generating immune-evading mutations, it is reasonable to assume that such infections are likely responsible for the emergence of at least some of the VOCs. This suggests that finding and treating individuals who are chronically infected must be a high public health priority165. Further, understanding the evolutionary dynamics of SARS-CoV-2 during chronic infections can provide a window into future potential emerging immune escape variants166.
Possible future scenarios
Until late 2021, before the emergence of Omicron, VOCs such as Alpha and Delta were mainly associated with increased transmissibility and modest degrees of immune escape167. However, current evidence suggests that immune escape properties were the main driver for the displacement of Delta by Omicron65. This implies that, over longer timescales, depending on the intrinsic transmissibility of future lineages and the degree of cross-immunity between them, one can imagine scenarios where two or more lineages co-circulate, or alternatively where one drives others to extinction. Such an interplay could have profound public health implications, depending on the transmissibility and virulence of the dominant lineage, and on the immunological landscape into which antigenically distinct lineages are introduced. For example, it is possible to envisage a scenario where waning immunity plus antigenic distinctiveness could periodically lead to new waves of infection.
A few studies have shown that some key, variant-defining mutations with ACE2-mediated transmissibility or resistance to population-level immunity could potentially be identified much earlier than the emergence of a new VOC98,168. However, many of these mutations may not pose a public health threat, unless in the presence of others. For example, the variant of interest Theta had a constellation of mutations that alerted scientists because it included defining mutations such as D614G, N501Y and E484K in the spike protein, yet its spread was limited. An effective early warning system for future VOCs requires rich information about variants, such as intrinsic transmissibility, tropism, immune escape, virulence and susceptibility to available treatments, linked to epidemiological data, such as the relative transmissibility of circulating variants and secondary attack rates from household transmission studies167,169. It may be possible to develop models that can predict some of these features from sequence data alone168,170. Wastewater surveillance provides a complementary source of sequence information171 and can reveal the cryptic transmission of variants in advance of their identification in individuals172. The role of immune escape mutations and their prediction will pose substantial challenges, as their fitness consequences depend on the immunity landscape. The Beta and Gamma VOCs for example, despite their local advantage, did not spread substantially outside the areas they were initially identified in, and Omicron BA.1 was outcompeted in animal models by both Alpha and Delta118.
With the caveats mentioned above, we can imagine a possible best-case scenario for the future evolution of SARS-CoV-2 whereby there will be continued antigenic drift within the Omicron lineage, such that over short and medium timescales, immunity elicited by a combination of vaccination and prior infection protects against severe disease on reinfection173,174, and provides broad immune responses that will cover considerable continued evolution of the virus175,176. In a best-case scenario, perhaps the majority of future fitness improving mutations will be limited to escape from host immunity. From the current trend of Omicron variants, we might expect a new wave of infection for every additional ~4 months of virus circulation136, although we have no way of knowing whether this periodicity would be maintained. As an illustration, if we imagined that SARS-CoV-2 infection fatality ratios in individuals with prior immunity were similar to those of seasonal influenza, we can expect two to three times the burden of influenza annually. This ignores any additional burden resulting from post-acute COVID-19 sequelae (also known as long COVID)177. Under this scenario, the longer-term impact of the virus would be determined by its levels of pathogenicity. The expectation is that it would also begin to follow a more regular seasonal incidence pattern, similar to other human coronaviruses178 (Box 2). The summer outbreaks of 2022 by Omicron-derived lineages hint that a regular seasonal pattern may not arrive soon.
A likely alternative to the best-case scenario is that antigenic evolution would be disrupted by the emergence of a new variant with a completely different constellation of mutations and phenotypic properties, which will allow the virus to evade immunity established by prior infection or vaccines. This could occur through accelerated evolution of the virus during long-term persistence within individuals who are immunocompromised who happen to carry a more basal strain of SARS-CoV-2 from a period when a completely different variant was circulating. A case in point is that both strains circulating in the white-tailed deer, the only known new reservoir in wildlife151, and a cryptic lineage found in urban wastewater161 feature pre-Omicron and even pre-VOC basal strains, demonstrating the continued persistence of basal strains. The unpredictability of the evolution of virulence could mean that such lineages are more virulent than Omicron, perhaps causing severe disease in more people. In such a case, the overall public health impact would be determined by the balance between severity of infections and residual population cross-immunity.
The emergence of VOCs and potential future antigenically distinct lineages can be thought of as ‘shift-like events’, which are unexpected, significant changes in the genetic make-up of the virus and, potentially, in its clinically relevant properties. Typical potential generators of shifts are recombinations. Although there is currently no evidence suggesting that recombination was involved in the origin of any VOCs, including Omicron65,179, recombination remains a continuing potential source of concern. Recombination events between highly divergent lineages have the capacity to bring potential adverse phenotypic properties together, for example, combining mutations that confer immune escape properties from one lineage with those that enhance transmissibility (and potentially also virulence) from another. Although likely not originating from recombination, the emergence events of each VOC bear the characteristics of a shift-like event. After what is believed to have been a long period of cryptic evolution, each VOC appeared unexpectedly, carried a large number of mutations and had considerably altered epidemiological or clinical characteristics. It is difficult to predict which part of viral genetic diversity future major lineages will originate from and whether they will result from ‘shift-like’ or more gradual, ‘drift-like’ evolution akin to that within the Omicron clade throughout 2022.
Other potential rare high-impact evolutionary events that could occur in the future could radically change the picture, but their likelihood is unquantifiable. These include, for example, recombination between SARS-CoV-2 and another virus, drastically altering the phenotype; spillback of divergent variants from animal reservoirs into humans; a complete change in the mode of transmission, akin to the one observed in the transmissible gastroenteritis virus, a coronavirus of animals180; and novel forms of vaccine escape that rapidly erode protection against severe disease and death.
In order to map out the repercussions of SARS-CoV-2 evolution for human health, we need to consider the intersection between its epidemiology and evolution. In the absence of eradication, the virus will likely become endemic, a process that could take years to decades178. We will be able to establish that endemic persistence has been reached if the virus shows repeatable patterns in prevalence year on year, for example, regular seasonal fluctuations and no out-of-season peaks. The form this endemic persistence will take remains to be determined181, and the eventual infection prevalence and disease burden will depend on the rate of emergence of antigenically distinct lineages, our ability to roll out and update vaccines, and the future trajectory of virulence (Fig. 4c). The tractability of the SARS-CoV-2 pandemic in the future will to a significant extent depend on the intensity of further evolutionary change, which in turn will depend on its global infection prevalence.
We have learned a great deal by studying the evolution of SARS-CoV-2 since its emergence in humans, and we can certainly make well-educated guesses about what is likely or unlikely to happen next, but the highly multifactorial and stochastic nature of the process will always keep the future evolutionary trajectory of the virus essentially unknown in many of its crucial details. Meanwhile, focusing on the epidemiology of the pathogen, it is important to bear in mind that the transition from a pandemic to future endemic existence of SARS-CoV-2 is likely to be long and erratic, rather than a short and distinct switch, and that endemic SARS-CoV-2 is by far not a synonym for safe infections, mild COVID-19 or a low population mortality and morbidity burden.
References
Khare, S. et al. GISAID’s role in pandemic response. China CDC Wkly 3, 1049–1051 (2021).
Pybus, O. G. & Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 10, 540–550 (2009).
Volz, E. et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell 184, 64–75.e11 (2021).
Clarke, D. K. et al. Genetic bottlenecks and population passages cause profound fitness differences in RNA viruses. J. Virol. 67, 222–228 (1993).
Sanjuán, R., Nebot, M. R., Chirico, N., Mansky, L. M. & Belshaw, R. Viral mutation rates. J. Virol. 84, 9733–9748 (2010).
Sanjuán, R. & Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 73, 4433–4448 (2016).
Loewe, L. & Hill, W. L. The population genetics of mutations: good, bad and indifferent. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 1153–1167 (2010).
Fehr, A. R. & Perlman, S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 1282, 1–23 (2015).
Amicone, M. et al. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 10, 142–155 (2022).
Minskaia, E., Hertzig, T., Gorbalenya, A. E. & Ziebuhr, J. Discovery of an RNA virus 3′→5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl Acad. Sci. USA 103, 5108–5113 (2006).
Ribeiro, R. M. et al. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog. 8, e1002880 (2012).
Rawson, J. M. O., Landman, S. R., Reilly, C. S. & Mansky, L. M. HIV-1 and HIV-2 exhibit similar mutation frequencies and spectra in the absence of G-to-A hypermutation. Retrovirology 12, 60 (2015).
Meng, B. et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 35, 109292 (2021).
Malim, M. H. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 675–687 (2009).
Jarmuz, A. et al. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79, 285–296 (2002).
Rogozin, I. B., Basu, M. K., Jordan, I. K., Pavlov, Y. I. & Koonin, E. V. APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predicted by computational analysis. Cell Cycle 4, 1281–1285 (2005).
Simmonds, P. & Ansari, M. A. Extensive C→U transition biases in the genomes of a wide range of mammalian RNA viruses; potential associations with transcriptional mutations, damage- or host-mediated editing of viral RNA. PLoS Pathog. 17, e1009596 (2021).
Klimczak, L. J., Randall, T. A., Saini, N., Li, J.-L. & Gordenin, D. A. Similarity between mutation spectra in hypermutated genomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 15, e0237689 (2020).
Kim, K. et al. The roles of APOBEC-mediated RNA editing in SARS-CoV-2 mutations, replication and fitness. Sci. Rep. 12, 14972 (2022).
Simmonds, P. Rampant C→U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses: causes and consequences for their short- and long-term evolutionary trajectories. mSphere 5, e00408–e00420 (2020).
Di Giorgio, S., Martignano, F., Torcia, M. G., Mattiuz, G. & Conticello, S. G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 6, eabb5813 (2020).
Ringlander, J., Fingal, J., Kann, H. & Kann, M. Impact of ADAR-induced editing of minor viral RNA populations on replication and transmission of SARS-CoV-2. Proc. Natl Acad. Sci. USA 119, e2112663119 (2022).
van Dorp, L. et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 83, 104351 (2020).
van Dorp, L. et al. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 11, 5986 (2020).
Belshaw, R., Sanjuán, R. & Pybus, O. G. Viral mutation and substitution: units and levels. Curr. Opin. Virol. 1, 430–435 (2011).
Rambaout, A. Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 16, 395–399 (2000).
Drummond, A. Nicholls, G. K., Rodrigo, A. G. & Solomon, W. in Tools for Constructing Chronologies: Crossing Disciplinary Boundaries Vol. 177 (eds Buck, C.E. & Maillard, A.R.) 149–171 (Springer-Verlag, 2004).
Duchene, S. et al. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 6, veaa061 (2020).
Ghafari, M. et al. Purifying selection determines the short-term time dependency of evolutionary rates in SARS-CoV-2 and pH1N1 influenza. Mol. Biol. Evol. 39, msac009 (2022).
Jackson, B. et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 184, 5179–5188 (2021).
Boni, M. F. et al. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417 (2020).
Lai, M. M. & Cavanagh, D. The molecular biology of corona viruses. Adv. Virus Res. 48, 1–100 (1997).
Rambaut, A. et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/1 (2020).
O’Toole, Á. et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 with Grinch. Wellcome Open Res. 6, 121 (2021).
Sekizuka, T. et al. Genome recombination between the Delta and Alpha variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Jpn J. Infect. Dis. 75, 415–418 (2022).
UK Health Security Agency (UKHSA). SARS-CoV-2 variants of concern and variants under investigation in England — Technical Briefing 39. GOV.UK https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1063424/Tech-Briefing-39-25March2022_FINAL.pdf (2022).
UK Health Security Agency (UKHSA). SARS-CoV-2 variants of public health interest: 28 October 2022. GOV.UK https://www.gov.uk/government/publications/sars-cov-2-variants-of-public-health-interest/sars-cov-2-variants-of-public-health-interest-28-october-2022 (2022).
Rhee, C., Kanjilal, S., Baker, M. & Klompas, M. Duration of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infectivity: when is it safe to discontinue isolation? Clin. Infect. Dis. 72, 1467–1474 (2021).
Bullard, J. et al. Predicting infectious severe acute respiratory syndrome coronavirus 2 from diagnostic samples. Clin. Infect. Dis. 71, 2663–2666 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Kissler, S. M. et al. Viral dynamics of SARS-CoV-2 variants in vaccinated and unvaccinated persons. N. Engl. J. Med. 385, 2489–2491 (2021).
Sun, K. et al. SARS-CoV-2 transmission, persistence of immunity, and estimates of Omicron’s impact in South African population cohorts. Sci. Transl Med. 14, eabo7081 (2022).
Cevik, M. et al. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. Lancet Microbe 2, e13–e22 (2021).
Hakki, S. et al. Onset and window of SARS-CoV-2 infectiousness and temporal correlation with symptom onset: a prospective, longitudinal, community cohort study. Lancet Respir. Med. 10, 1061–1073 (2022).
Lythgoe, K. A. et al. SARS-CoV-2 within-host diversity and transmission. Science 372, eabg0821 (2021).
Ke, R. et al. Daily longitudinal sampling of SARS-CoV-2 infection reveals substantial heterogeneity in infectiousness. Nat. Microbiol. 7, 640–652 (2022).
Farjo, M. et al. Within-host evolutionary dynamics and tissue compartmentalization during acute SARS-CoV-2 infection. Preprint at bioRxiv https://doi.org/10.1101/2022.06.21.497047 (2022).
Martin, M. A. & Koelle, K. Comment on ‘Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2’. Sci. Transl Med. 13, eabh1803 (2021).
Koelle, K. et al. Masks do not more than prevent transmission: theory and data undermine the variolation hypothesis. Preprint at medRxiv https://doi.org/10.1101/2022.06.28.22277028 (2022).
Lumby, C. K., Nene, N. R. & Illingworth, C. J. R. A novel framework for inferring parameters on transmission from viral sequence data. PLoS Genet. 14, e1007718 (2018).
Zwart, M. P. & Elena, S. F. Matters of size: genetic bottlenecks in virus infection and their potential impact on evolution. Annu. Rev. Virol. 2, 161–179 (2015).
McCrone, J. T. et al. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 7, 35962 (2018).
Sobel Leonard, A., Weissman, D. B., Greenbaum, B., Ghedin, E. & Koelle, K. Transmission bottleneck size estimation from pathogen deep-sequencing data, with an application to human influenza A virus. J. Virol. 91, 00171-17 (2017).
Ghafari, M., Lumpy, C. K., Weissman, D. B. & Illingworth, C. J. R. Inferring transmission bottleneck size from viral sequence data using a novel haplotype reconstruction method. J. Virol. 94, e00014–e00020 (2020).
Joseph, S. B., Swanstrom, R., Kashuba, A. D. M. & Cohen, M. S. Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nat. Rev. Microbiol. 13, 414–425 (2015).
Gutiérrez, S., Michalakis, Y. & Blanc, S. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2, 546–555 (2012).
Adam, D. C. et al. Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong. Nat. Med. 26, 1714–1719 (2020).
Liu, Y., Eggo, R. M. & Kucharski, A. J. Secondary attack rate and superspreading events for SARS-CoV-2. Lancet 395, e47 (2020).
Wright, S. Evolution in Mendelian populations. Genetics 16, 97–159 (1931).
Grubaugh, N. D., Hanage, W. P. & Rasmussen, A. L. Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell 182, 794–795 (2020).
Korber, B. et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827.e19 (2020).
Hou, Y. J. et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 370, 1464–1468 (2020).
Meyer, A. G., Spielman, S. J., Bedford, T. & Wilke, C. O. Time dependence of evolutionary metrics during the 2009 pandemic influenza virus outbreak. Virus Evol. 1, vev006 (2015).
Holmes, E. C., Dudas, G., Rambaut, A. & Andersen, K. G. The evolution of Ebola virus: insights from the 2013–2016 epidemic. Nature 538, 193–200 (2016).
Viana, R. et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603, 679–686 (2022).
Lythgoe, K. A. et al. Lineage replacement and evolution captured by the United Kingdom Covid Infection Survey. Preprint at medRxiv https://doi.org/10.1101/2022.01.05.21268323 (2022).
Tay, J. H., Porter, A. F., Wirth, W. & Duchene, S. The emergence of SARS-CoV-2 variants of concern is driven by acceleration of the substitution rate. Mol. Biol. Evol. 39, msac013 (2022).
Gräf, T. et al. Identification of a novel SARS-CoV-2 P.1 sub-lineage in Brazil provides new insights about the mechanisms of emergence of variants of concern. Virus Evol. 7, veab091 (2021).
Neher, R. A. Contributions of adaptation and purifying selection of SARS-CoV-2 evolution. Virus Evol. 8, veac113 (2022).
Saito, A. et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75 variant. Cell Host Microbe 30, 1540–1555 (2022).
Ito, J. et al. Convergent evolution of the SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Preprint at bioRxiv https://doi.org/10.1101/2022.12.05.519085 (2022).
Sousa, W. P. & Grosholz, E. D. in Habitat Structure Vol. 8 (eds Bell, S. S., McCoy, E. D. & Mushinsky, H. R.) 300–324 (Springer, 1991).
Hilleman, M. R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc. Natl Acad. Sci. USA 101, 14560–14566 (2004).
Domingo, E. in Virus as Populations Ch. 5 (ed. Domingo, E.) 167–194 (Academic, 2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Sinha, S., Tam, B. & Ming Wang, S. RBD double mutations of SARS-CoV-2 strains increase transmissibility through enhanced interaction between RBD and ACE2 receptor. Viruses 14, 1 (2022).
Yurkovetskiy, L. et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell 183, 739–751 (2020).
Ozono, S. et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 12, 848 (2021).
Liu, H. et al. The basis of a more contagious 501Y.V1 variant of SARS-CoV-2. Cell Res. 31, 720–722 (2021).
Benton, D. J. et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 588, 327–330 (2020).
Peacock, T. P. et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 6, 899–909 (2021).
Wrobel, A. G. et al. Evolution of the SARS-CoV-2 spike protein in the human host. Nat. Commun. 13, 1178 (2022).
Johnson, B. A. et al. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. PLoS Pathog. 18, e1010627 (2022).
Thorne, L. G. et al. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 602, 487–495 (2022).
Lamers, M. M. et al. SARS-CoV-2 Omicron efficiently infects human airway, but not alveolar epithelium. Preprint at bioRxiv https://doi.org/10.1101/2022.01.19.476898 (2022).
Hui, K. P. Y. et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 603, 715–720 (2022).
Port, J. et al. Increased small particle aerosol transmission of B.1.1.7 compared with SARS-CoV-2 lineage A in vivo. Nat. Microbiol. 7, 213–223 (2022).
Bushmaker, T. et al. Comparative aerosol and surface stability of SARS-CoV-2 variants of concern. Preprint at bioRxiv https://doi.org/10.1101/2022.11.21.517352 (2022).
Oswin, H. P., Haddrell, A. E., Otern-Fernandez, M. & Reid, J. P. The dynamics of SARS-CoV-2 infectivity with changes in aerosol microenvironment. Proc. Natl Acad. Sci. USA 119, e2200109119 (2022).
King, A. A., Schresta, S., Harvill, E. T. & Bjornstad, O. N. Evolution of acute infections and the invasion‐persistence trade‐off. Am. Nat. 173, 446–455 (2009).
Lehtinen, S., Ashcroft, P. & Bonhoeffer, S. On the relationship between serial interval, infectiousness profile and generation time. J. R. Soc. Interface 18, 20200756 (2021).
Wallinga, J. & Lipsitch, M. How generation intervals shape the relationship between growth rates and reproductive numbers. Proc. Biol. Sci. 274, 599–604 (2007).
Backer, J. A. et al. Shorter serial intervals in SARS-CoV-2 cases with Omicron BA.1 variant compared with Delta variant, the Netherlands, 13 to 26 December 2021. Eur. Surveill. 27, 2200042 (2022).
Hay, J. A. et al. Quantifying the impact of immune history and variant on SARS-CoV-2 viral kinetics and1 infection rebound: a retrospective cohort study. eLife 11, e81849 (2022).
Pulliam, J. R. et al. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science 376, 596 (2022).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Starr, T. N. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182, 1295–1310.e20 (2020).
Greaney, A. J. et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 29, 44–57.e9 (2021).
Dejnirattisai, W. et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 185, 467–484.e15 (2022).
McCallum, M. et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 375, 864–868 (2022).
Nutalai, R. et al. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 185, 2116–2131.e18 (2022).
Tuekprakhon, A. et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 185, 2422–2433.e13 (2022).
Kistler, K. E., Huddleston, J. & Bedford, T. Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2. Cell Host Microbe 30, 545–555.e4 (2022).
Obermeyer, F. et al. Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 376, 1327–1332 (2022).
Naranbhai, V. et al. T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals. Cell 185, 1041–1051 (2022).
Yu, F., Tai, W. & Cheng, G. T-cell immunity: a barrier to Omicron immune evasion. Sig. Transduct. Target. Ther. 7, 297 (2022).
Riu, C. et al. Escape from recognition of SARS-CoV-2 variant spike epitopes but overall preservation of T cell immunity. Sci. Transl Med. 14, eabj6824 (2022).
Dolton, G. et al. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell 185, 2936–2951 (2022).
Agerer, B. et al. SARS-CoV-2 mutations in MHC-I-restricted epitopes evade CD8+ T cell responses. Sci. Immunol. 6, eabg646 (2021).
Chang, M. R. et al. Analysis of a SARS-CoV-2 convalescent cohort identified a common strategy for escape of vaccine-induced anti-RBD antibodies by Beta and Omicron variants. eBioMedicine 80, 104025 (2022).
Tada, T. et al. Partial resistance of SARS-CoV-2 Delta variants to vaccine-elicited antibodies and convalescent sera. iScience 24, 103341 (2021).
Reed, A. F. The evolution of virulence. Trends Microbiol. 2, 73–76 (1994).
Tegally, H. et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592, 438–443 (2021).
Funk, T. et al. Characteristics of SARS-CoV-2 variants of concern B.1.1.7, B.1.351 or P.1: data from seven EU/EEA countries, weeks 38/2020 to 10/2021. Eur. Surveill. 26, 2100348 (2021).
Public Health England. SARS-CoV-2 variants of concern and variants under investigation in England Technical briefing 16 2021. GOV.UK https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/997414/Variants_of_Concern_VOC_Technical_Briefing_16.pdf (2021).
European Centre for Disease Prevention and Control. Assessment of the further spread and potential impact of the SARS-CoV-2 Omicron variant of concern in the EU/EEA, 19th update. ECDC https://www.ecdc.europa.eu/en/publications-data/covid-19-omicron-risk-assessment-further-emergence-and-potential-impact (2022).
Barut, G. T. et al. The spike gene is a major determinant for the SARS-CoV-2 Omicron-BA.1 phenotype. Nat. Commun. 13, 5929 (2022).
Chen, D.-Y. et al. Spike and nsp6 are key determinants of SARS-CoV-2 Omicron BA.1 attenuation. Nature 615, 143–150 (2023).
Liu, S., Selvaraj, P., Sangare, K., Luan, B. & Wang, T. T. Spike protein-independent attenuation of SARS-CoV-2 Omicron variant in laboratory mice. Cell Rep. 40, 111359 (2022).
Markov, P. V., Katzourakis, A. & Stilianakis, N. I. Antigenic evolution will lead to new SARS-CoV-2 variants with unpredictable severity. Nat. Rev. Microbiol. 20, 251–252 (2022).
Elsworth, P. et al. Increased virulence of rabbit haemorrhagic disease virus associated with genetic resistance in wild Australian rabbits (Oryctolagus cuniculus). Virology 464–465, 415–423 (2014).
Lange, M. & Thulke, H.-H. Elucidating transmission parameters of African swine fever through wild boar carcasses by combining spatio-temporal notification data and agent-based modelling. Stoch. Environ. Res. Risk Assess. 31, 379–391 (2017).
World Health Organization (WHO). WHO Middle East respiratory syndrome: global summary and assessment of risk — 16 November 2022. WHO https://www.who.int/publications/i/item/WHO-MERS-RA-2022.1 (2022).
COVID-19 excess mortality collaborators. Estimating excess mortality due to the COVID-19 pandemic: a systematic analysis of COVID-19-related mortality, 2020–21. Lancet 399, 1513–1536 (2022).
Davies, N. G. et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 372, eabg3055 (2021).
Shen, X. et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe 29, 529–539.e3 (2021).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Konings, F. et al. SARS-CoV-2 variants of interest and concern naming scheme conducive for global discourse. Nat. Microbiol. 6, 821–823 (2021).
Faria, N. R. et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 372, 815–821 (2021).
Zhou, D. et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 184, 2348–2361.e6 (2021).
Sabino, E. C. et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet 397, 452–455 (2021).
Dhar, M. S. et al. Genomic characterization and epidemiology of an emerging SARS-CoV-2 variant in Delhi, India. Science 374, 995–999 (2021).
Bolze, A. et al. SARS-CoV-2 variant Delta rapidly displaced variant Alpha in the United States and led to higher viral loads. Cell Rep. Med. 3, 100564 (2022).
Campbell, F. et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eur. Surveill. 26, 2100509 (2021).
Tegally, H. et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 28, 1785–1790 (2022).
Attwood, S. W., Hill, S. C., Aanensen, D. M., Connor, T. R. & Pybus, O. G. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 23, 547–562 (2022).
Tao, K. et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 22, 757–773 (2021).
Hill, V. et al. The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK. Virus Evol. 8, veac080 (2022).
McCrone, J. T. et al. Context-specific emergence and growth of the SARS-CoV-2 Delta variant. Nature 610, 154–160 (2022).
Adepoju, P. Challenges of SARS-CoV-2 genomic surveillance in Africa. Lancet Microbe 2, e139 (2021).
Wilkinson, E. et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 374, 423–431 (2021).
Mandolo, J. et al. SARS-CoV-2 exposure in Malawian blood donors: an analysis of seroprevalence and variant dynamics between January 2020 and July 2021. BMC Med. 19, 303 (2021).
Ghafari, M., Watson, O. J., Karlinsky, A., Ferretti, L. & Katzourakis, A. A framework for reconstructing SARS-CoV-2 transmission dynamics using excess mortality data. Nat. Commun. 13, 3015 (2022).
Ghafari, M., Liu, Q., Dhillon, A., Katzourakis, A. & Weissman, D. B. Investigating the evolutionary origins of the first three SARS-CoV-2 variants of concern. Front. Virol. 2, 942555 (2022).
Schlottau, K. et al. SARS-CoV-2 in fruit bats, ferrets, pigs, and chickens: an experimental transmission study. Lancet Microbe 1, e218–e225 (2020).
Muñoz-Fontela, C. et al. Advances and gaps in SARS-CoV-2 infection models. PLoS Pathog. 18, e1010161 (2022).
Oude Munnink, B. B. et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 371, 172–177 (2021).
Hale, V. L. et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature 602, 481–486 (2022).
Ren, W. et al. Mutation Y453F in the spike protein of SARS-CoV-2 enhances interaction with the mink ACE2 receptor for host adaption. PLoS Pathog. 17, e1010053 (2021).
Pickering, B. et al. Divergent SARS-CoV-2 variant emerges in white-tailed deer with deer-to-human transmission. Nat. Microbiol. 7, 2011–2024 (2022).
Porter, A. F., Purcell, D. F. J., Howden, B. P. & Duchene, S. Evolutionary rate of SARS-CoV-2 increases during zoonotic infection of farmed mink. Virus Evol. 9, vead002 (2023).
Diamond, M. et al. The SARS-CoV-2 B.1.1.529 Omicron virus causes attenuated infection and disease in mice and hamsters. Preprint at Res. Sq. https://doi.org/10.21203/rs.3.rs-1211792/v1 (2021).
Shuai, H. et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 603, 693–699 (2022).
Dinnon, K. H. III et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020).
Weigang, S. et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 12, 6405 (2021).
Choi, B., Choudhary, M. C., Regan, J., Sparks, J. A. & Padera, R. F. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 383, 2291–2293 (2020).
Clark, S. A. et al. SARS-CoV-2 evolution in an immunocompromised host reveals shared neutralization escape mechanisms. Cell 184, 2605–2617.e18 (2021).
Msomi, N., Lessells, R., Mlisana, K. & de Oliveira, T. Africa: tackle HIV and COVID-19 together. Nature 600, 33–36 (2021).
Wilkinson, S. A. J. et al. Recurrent SARS-CoV-2 mutations in immunodeficient patients. Virus Evol. 8, veac050 (2022).
Gregory, D. A. et al. Genetic diversity and evolutionary convergence of cryptic SARS- CoV-2 lineages detected via wastewater sequencing. PLoS Pathog. 18, e1010636 (2022).
Gonzalez-Reiche, A. S. et al. SARS-CoV-2 variants in the making: sequential intrahost evolution and forward transmissions in the context of persistent infections. Preprint at bioRxiv https://doi.org/10.1101/2022.05.25.22275533 (2022).
Harari, S. et al. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nat. Med. 28, 1501–1508 (2022).
Moran, E. et al. Persistent SARS-CoV-2 infection: the urgent need for access to treatment and trials. Lancet Infect. Dis. 21, 1345–1347 (2021).
Dennehy, J. J., Gupta, R. K., Hanage, W. P., Johnson, M. C. & Peacock, T. P. Where is the next SARS-CoV-2 variant of concern? Lancet 399, 1938–1939 (2022).
Lemieux, J. E. & Luban, J. Consulting the Oracle of SARS-CoV-2 infection. J. Infec. Dis. 225, 1115–1117 (2022).
Oude Munnink, B. B. et al. The next phase of SARS-CoV-2 surveillance: real-time molecular epidemiology. Nat. Med. 27, 1518–1524 (2021).
Maher, M. C. et al. Predicting the mutational drivers of future SARS-CoV-2 variants of concern. Sci. Transl Med. 14, eabk3445 (2022).
Subissi, L. et al. An early warning system for emerging SARS-CoV-2 variants. Nat. Med. 28, 1110–1115 (2022).
Telenti, A., Hodcroft, E. B. & Robertson, D. L. The evolution and biology of SARS-CoV-2 variants. Cold Spring Harb. Perspect. Med. 12, a041390 (2022).
Amman, F. et al. Viral variant-resolved wastewater surveillance of SARS-CoV-2 at national scale. Nat. Biotechnol. 40, 1814–1822 (2022).
Karthikeyan, S. et al. Wastewater sequencing reveals early cryptic SARS-CoV-2 variant transmission. Nature 609, 101–108 (2022).
Gao, Y. et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat. Med. 28, 472–476 (2022).
Keeton, R. et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 603, 488–492 (2022).
Kitchin, D. et al. Ad26.COV2.S breakthrough infections induce high titers of neutralizing antibodies against Omicron and other SARS-CoV-2 variants of concern. Cell Rep. Med. 3, 100535 (2022).
He, W.-T. et al. Targeted isolation of diverse human protective broadly neutralizing antibodies against SARS-like viruses. Nat. Immunol. 23, 960–970 (2022).
Al-Aly, Z., Bowe, B. & Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat. Med. 28, 1461–1467 (2022).
Lavine, J. S., Bjornstad, O. N. & Antia, R. Immunological characteristics govern the transition of COVID-19 to endemicity. Science 371, 741–745 (2021).
Calaway, E. Heavily mutated Omictorn varaints puts scientists into alert. Nature 600, 21 (2021).
Pensaert, M., Callebaut, P. & Vergote, J. Isolation of a porcine respiratory, non-enteric coronavirus related to transmissible gastroenteritis. Vet. Q. 8, 257–261 (1986).
Katzourakis, A. COVID-19: endemic doesn’t mean harmless. Nature 601, 485 (2022).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Stilianakis, N. I., Perelson, A. S. & Hayden, F. G. Emergence of drug resistance during an influenza epidemic: insights from a mathematical model. J. Infect. Dis. 177, 863–873 (1998).
Clavel, F. & Hance, A. J. HIV drug resistance. N. Engl. J. Med. 350, 1023–1035 (2004).
Holmes, E. C. et al. Understanding the impact of resistance to influenza antivirals. Clin. Microbiol. Rev. 34, e00224-20 (2021).
Artese, A. et al. Current status of antivirals and druggable targets of SARS CoV-2 and other human pathogenic coronaviruses. Drug Resist. Updat. 53, 100721 (2020).
Hussain, M., Galvin, H. D., Haw, T. Y., Nutsford, A. N. & Husain, M. Drug resistance in influenza A virus: the epidemiology and management. Infect. Drug Resist. 10, 121–134 (2017).
Perelson, A. S., Rong, L. & Hayden, F. G. Combination antiviral therapy for influenza: predictions from modeling of human infections. J. Infect. Dis. 205, 1642–1645 (2012).
Dunning, J., Baillie, J. K., Cao, B. & Hayden, F. G. International severe acute respiratory and emerging infection consortium (ISARIC). Antiviral combinations for severe influenza. Lancet Infect. Dis. 14, 1259–1270 (2014).
Hammond, J. et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N. Engl. J. Med. 386, 1397–1408 (2022).
Jeong, J. H. et al. Combination therapy with nirmatrelvir and molnupiravir improves the survival of SARS-CoV-2 infected mice. Antivir. Res. 208, 105430 (2022).
National Institues of Health (NIH). Antiviral agents, including antibody products. NIH.GOV https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/summary-recommendations/ (2023).
Szemiel, A. M. et al. In vitro selection of remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 17, e1009929 (2021).
Stevens, L. J. et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl Med. 14, eabo0718 (2022).
Zhou, Y. et al. Nirmatrelvir resistant SARS-CoV-2 variants with high fitness in vitro. Sci. Adv. 8, eadd7197 (2022).
Malone, B. & Campbell, E. A. Molnupiravir: coding for catastrophe. Nat. Struct. Mol. Biol. 28, 706–708 (2021).
Pillai, S. K., Wong, J. K. & Barbour, J. D. Turning up the volume on mutational pressure: is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3). Retrovirology 5, 26 (2008).
Donovan-Banfield, I. et al. Characterisation of SARS-CoV-2 genomic variation in response to molnupiravir treatment in the AGILE phase IIa clinical trial. Nat. Commun. 13, 7284 (2022).
Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Ansino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Pfeiffer, J. K. & Kirkegaard, K. Increased fidelity reduces poliovirus fitness and virulence under selective pressure in mice. PLoS Pathog. 1, e11 (2005).
Sanderson, T., Hisner, R., Donovan-Banfield, I., Peackock, T. & Ruis, C. Identification of a molnupiravir-associated mutational signature in SARS-CoV-2 sequencing databases. Preprint at medRxiv https://doi.org/10.1101/2023.01.26.23284998 (2023).
Hoffmann, M. et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 184, 2384–2393.e12 (2021).
Focosi, D. et al. Monoclonal antibody therapies against SARS-CoV-2. Lancet Infect. Dis. 22, e311–e326 (2022).
Choudhary, M. C. et al. Emergence of SARS-CoV-2 escape mutations during Bamlanivimab therapy in a phase II randomized clinical trial. Nat. Microbiol. 7, 1906–1917 (2022).
Gottlieb, R. L. et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA 325, 632–644 (2021).
Greaney, A. J. et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 12, 4196 (2021).
Chen, R. E. et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 27, 717–726 (2021).
Corman, V. M., Muth, D., Niemeyer, D. & Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 100, 163–188 (2018).
Cui, J., Li, F. & Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192 (2019).
Cheng, V. C. C., Lau, S. K. P., Woo, P. C. Y. & Yuen, K. Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 20, 660–694 (2007).
Kiyuka, P. K. et al. Human coronavirus NL63 molecular epidemiology and evolutionary patterns in rural coastal Kenya. J. Infect. Dis. 217, 1728–1739 (2018).
Larson, H. E., Reed, S. E. & Tyrrell, D. A. Isolation of rhinoviruses and coronaviruses from 38 colds in adults. J. Med. Virol. 5, 221–229 (1980).
Vijgen, L. et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79, 1595–1604 (2005).
Pollett, S. et al. A comparative recombination analysis of human coronaviruses and implications for the SARS-CoV-2 pandemic. Sci. Rep. 11, 17365 (2021).
Akaishi, T. Insertion-and-deletion mutations between the genomes of SARS-CoV, SARS-CoV-2, and bat coronavirus RaTG13. Microbiol. Spectr. 10, e0071622 (2022).
Coutard, B. et al. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 176, 104742 (2020).
Ren, W. et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 82, 1899–1907 (2008).
Guo, H. et al. Identification of a novel lineage bat SARS-related coronaviruses that use bat ACE2 receptor. Emerg. Microbes Infect. 10, 1507–1514 (2021).
Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004).
Acknowledgements
A.K. was supported by the European Research Council (ERC) Consolidator Grant PALVIREVOL-101001623.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article. P.V.M. and A.K. contributed substantially to discussion of the content. P.V.M., M.G., M.B., P.S, N.I.S. and A.K. wrote the article. P.V.M., M.G. and A.K. reviewed and/or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Microbiology thanks Thomas Connor, Robert Garry and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Disclaimer
The views expressed in this article are purely those of the authors and may not, under any circumstances, be regarded as an official position of the European Commission.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Nextstrain: https://nextstrain.org/
Glossary
- Antibody neutralization
-
The blocking of viral replication in a host as a result of antibody binding to the virus. Often this is due to binding of the antibody to viral structures involved in interactions with cellular receptors, resulting in blocking of cell entry.
- Basic reproduction number
-
(R0). The average total number of secondary infections that a single infectious case produces in a totally susceptible population of hosts. A measure of transmissibility.
- Consensus sequence
-
The sequence obtained by taking the most common base at each nucleotide position along the genome from the often genetically diverse population in a sample.
- Duration of infectiousness
-
The duration of the period in which a host is infectious with a specific pathogen. This can vary from a few days to decades or even a lifetime in different pathogens.
- Effective population size
-
The size of an idealized population, which determines the changes in gene frequency as a result of genetic drift and the effectiveness of selection relative to drift. An intuitive explanation of this abstract term is that the effective population size approximates the number of individuals in a population who are actually reproducing and leaving progeny.
- Endemic persistence
-
An epidemiological state where a pathogen exists at long-term stable prevalence in the host population, neither increasing nor decreasing. Endemic persistence implies neither low virulence nor low prevalence of the pathogen, but these are often incorrectly assumed attributes of the term, confusing debates on the subject.
- Fitness
-
The reproductive success of an organism. For viruses spreading in a host population, fitness is often approximated by the net reproduction number (Rt) — the number of new infections with a viral variant a single infectious person produces in the population.
- Founder effect
-
The loss of genetic diversity in the population as a result of a new population being established by a small number of individuals. It is a consequence of the transmission bottleneck, significantly influenced by chance and contributes to genetic drift.
- Genetic drift
-
Changes in a nucleotide character frequency at a genomic site in a population over time due to chance. Drift enables some characters to become common despite a lack of selective advantage, or even if mildly deleterious.
- Immune escape
-
The ability of a virus to partially or fully evade immune recognition or neutralization. Here we only focus on mutational immune escape, which results from virus evolution.
- Infection fatality ratio
-
The ratio of the number of deaths caused by a pathogen infection to the number of individuals infected by the pathogen. It is a measure of the infection severity and the virulence of the pathogen (often also called the ‘infection fatality rate’, despite not being a rate).
- Infectiousness
-
The intensity with which a pathogen is expelled by an infectious host, usually as a result of high infectious loads, infectiousness promoting symptoms and presence in tissues or fluids, which are important in its transmission route.
- Infectivity
-
The ability of a pathogen to take foothold in a new host and establish infection.
- Intrinsic transmissibility
-
The capacity of an infectious agent to move successfully from an infectious host to a susceptible one, successfully establishing infection in it. This includes the intensity with which it is expelled by the infectious host, its ability to resist factors in the environment while in transit and its ability to establish in a new susceptible host.
- Latent period
-
The duration between the moment of infection of an individual host and the moment that host starts to be infectious to others; characterizes infectious agents.
- Most recent common ancestor
-
The most recent individual from which a group of individuals in the population are descended.
- Mutation rate
-
The probability of mutation, usually measured per nucleotide per replication cycle.
- Mutations
-
Changes in the genome of the virus, usually occurring as a result of errors during replication.
- Net reproduction number
-
(Rt). The average total number of secondary infections that a single infectious case produces in a real population of hosts, where epidemiological control measures may be applied and a proportion of the population may be immune.
- Recombination
-
The combining of genetic material from two different viruses during replication, producing an offspring virus carrying a portion of the genetic material from either parent.
- Severity
-
The gravity of a clinical condition or infection.
- Substitution rate
-
(Also known as evolutionary rate). The rate at which new mutations accumulate in a viral population, usually measured per nucleotide site per year.
- Superspreading
-
A situation whereby a particular setting, circumstances or an infectious individual are responsible for the transmission of a pathogen to a disproportionately large number of people.
- Transmissibility
-
The intensity with which an infectious agent moves between hosts in a real setting, where hosts may have prior immunity. Besides intrinsic transmissibility of the pathogen, this includes immune evasion as well as temporal components such as duration and time of onset of infectiousness.
- Transmission bottleneck
-
The infinitesimal number of viral particles that establish the viral population in a new host on transmission. Usually, these are a minuscule and often genetically unrepresentative sample from the virus population in the original host, which contributes to genetic drift.
- Variant of interest
-
As defined by the World Health Organization, a severe acute respiratory syndrome coronavirus 2 variant with genetic changes predicted or known to affect its transmissibility, disease severity, immune escape, or diagnostic or therapeutic escape, causing significant community transmission, in multiple countries, increasing in frequency, causing increasing number of cases and suggesting an emerging risk to global health.
- Variants of concern
-
(VOCs). Variants that, in addition to the criteria for a variant of interest listed above, are associated with an increase in transmissibility, an increase in virulence or a decrease in effectiveness of public health measures, diagnostics, vaccines and therapeutics at a degree of global health significance.
- Virulence
-
Casually defined as the degree of harm a pathogen causes to a host.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Markov, P.V., Ghafari, M., Beer, M. et al. The evolution of SARS-CoV-2. Nat Rev Microbiol 21, 361–379 (2023). https://doi.org/10.1038/s41579-023-00878-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41579-023-00878-2
This article is cited by
-
Evaluation of the implementation of a community health worker-led COVID-19 contact tracing intervention in Chiapas, Mexico, from March 2020 to December 2021
BMC Health Services Research (2024)
-
Effectiveness and safety of azvudine in older adults with mild and moderate COVID-19: a retrospective observational study
BMC Infectious Diseases (2024)
-
Quality control in SARS-CoV-2 RBD-Fc vaccine production using LC–MS to confirm strain selection and detect contaminations from other strains
Scientific Reports (2024)
-
Network analysis-guided drug repurposing strategies targeting LPAR receptor in the interplay of COVID, Alzheimer’s, and diabetes
Scientific Reports (2024)
-
Risk for diagnosis or treatment of mood or anxiety disorders in adults after SARS-CoV-2 infection, 2020–2022
Molecular Psychiatry (2024)
