
Abstract
Yellow-cheek carp (Elopichthys bambusa) is a typical large and ferocious carnivorous fish endemic to East Asia, with high growth rate, nutritional value and economic value. In this study, a chromosome-level genome of yellow-cheek carp was generated by combining PacBio reads, Illumina reads and Hi-C data. The genome size is 827.63 Mb with a scaffold N50 size of 33.65 Mb, and 99.51% (823.61 Mb) of the assembled sequences were anchored to 24 pseudo-chromosomes. The genome is predicted to contain 24,153 protein-coding genes, with 95.54% having functional annotations. Repeat elements account for approximately 55.17% of the genomic landscape. The completeness of yellow-cheek carp genome assembly is highlighted by a BUSCO score of 98.4%. This genome will help us understand the genetic diversity of yellow-cheek carp and facilitate its conservation planning.
Similar content being viewed by others
Background & Summary
Yellow-cheek carp (Elopichthys bambusa), also known as “water tiger”, is a species in the order Elopichthys, subfamily Leuciscinae and family Cyprinidae. Yellow-cheek carp is a typical large and ferocious carnivorous fish endemic to East Asia. In China, it is mainly distributed in river systems such as the Yangtze River, Pearl River and Yellow River1. Yellow-cheek carp lives in the upper layer of rivers and lakes, it has a strong swimming ability and chases other fish for food. Yellow-cheek carp can prey on diseased and weak fish to control their population size, which is of great significance for maintaining the ecological balance of the water environment2. Yellow-cheek carp is also an important characteristic economic fish with firm meat, delicious taste, and rich in high-quality protein, unsaturated fatty acids, minerals and other nutrients3,4,5. However, anthropic factors such as overfishing, hydrological modification and water pollution have led to the dwindling natural resources of yellow-cheek carp6,7, which has been listed in the “Key Protected Endangered and Threatened Aquatic Species” and the IUCN Red List of Threatened Species (Version 2020.3)8.
The typical carnivorous yellow-cheek carp is particularly special among East Asian carp species that are mainly omnivorous and herbivorous. For example, yellow-cheeked carp and grass carp both belong to the subfamily Leuciscinae and had the closest relationship. Interestingly, they have evolved completely opposite feeding habits9, which provides excellent material for studying the evolution and genetic regulation mechanisms of fish feeding habits. However, the lack of genomic information limits the study on the carnivorous formation mechanism of yellow-cheek carp. At the same time, higher breeding profits have also promoted the continuous development of the artificial breeding industry of yellow-cheek carp. Using live fish or frozen fish as the main bait not only results in higher breeding costs for yellow-cheeked carp, but also easily causes pollution of the aquaculture water, which greatly restricts the expansion of the farming scale10. Therefore, research on the dietary transformation of typical carnivorous fishes such as yellow-cheek carp has gradually become a hot topic, and there is an urgent need for genetic breeding of yellow-cheek carp based on whole-genome information.
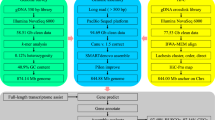
In this research, we have combined PacBio long-read sequencing, Illumina short-read sequencing and Hi-C technology to generate a high-quality chromosome-level genome of the yellow-cheek carp (Fig. 1). Accordingly, we expect rapid progress in the genetics research of yellow-cheeked carp, and functional genes related to key economic traits of yellow-cheeked carp will continue to be discovered. The elucidation of the genome structures and functions will promote more in-depth research to better understand the genetic basis for the formation of important traits such as the carnivorous in yellow-cheeked carp, thereby making contributions to its resource protection, genetic selection and artificial breeding.

Characterization of assembled yellow-cheek carp genome. Circos plot of the yellow-cheek carp genome, with visualization of gene density (1), TRP (2), LTR (3), SINE (4), LINE (5) and GC content (6) in order from outside to inside.
Methods
Sample collection and sequencing
An adult male yellow-cheek carp was collected from the Yangtze River in Wuhan, Hubei, China. High-quality genomic DNA was extracted from muscle by the CTAB method for Illumina sequencing, PacBio SMRT sequencing11 and Hi-C. The quality of the extracted DNA was assessed using agarose gel electrophoresis and NanoDrop Spectrophotometer (Thermo Fisher Scientific, USA), and quantified by a Qubit Fluorometer (Invitrogen, USA).
For Illumina sequencing, the genomic DNA was randomly sheared to 300~500 bp fragments, and a paired-end genomic library was prepared following the manufacturer’s protocol. Then, the library was sequenced on an Illumina NovaSeq platform using a paired-end 150 bp layout to enable genome survey and base-level correction. For PacBio long-read sequencing, SMRTbell libraries were constructed using the genomic DNA and sequenced on the PacBio Sequel II sequencing platform. After, approximately 58.98 Gb of Illumina short-read data (coverage of 71.31×) and 27.35 Gb of PacBio continuous long reads (CLR) data (coverage of 32.65×) was obtained.
To generate a chromosomal-level assembly of the yellow-cheek carp genome, a Hi-C library was generated using the DNA extracted from the same yellow-cheek carp. After cell crosslinking, cell lysis, chromatin digestion, biotin labelling, proximal chromatin DNA ligation and DNA purification, the resulting Hi-C library was subjected to paired-end sequencing with 150 bp read lengths on an Illumina NovaSeq platform. Finally, the size of Hi-C data obtained was 151.98 Gb, covering 183.78× of the genome.
To aid genome annotation, the total RNA from muscle, spleen, gonad and skin was extracted and tested for purity and integrity using a NanoDrop Spectrophotometer (Thermo Fisher Scientific, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, USA). The RNA library was constructed using the NEBNext® UltraTM RNA Library Prep Kit (Illumina, USA) following the manufacturer’s protocol and sequenced on an Illumina NovaSeq. 6000 platform. Finally, 23.74 Gb of data was obtained (Table 1).
Genome assembly
First, SOAPnuke (v2.1.0)12 was used to perform quality control of Illumina data, and the clean data were utilized for genome size estimation. K-mer analysis13 was conducted using GCE (v1.0.2). As a result, the genome size was estimated to be 786.16 Mb, with a heterozygosity ratio of 0.47% and repeat sequence ratio of 47.03% (Table 2). A total of 27.35 Gb PacBio long-read data were used for de novo genome assembly using MECAT2 (v2.0.0)14 and NextDenovo (v2.4.0). The polishing was then carried out by the software gcpp (v2.0.2) and pilon (v1.22)15. Based on these sequencing data, the resulting assembly consists of 170 contigs and has a total length of 827.63 Mb (Table 3).
Hi-C scaffolding
The Hi-C technology was used for chromosome-level genome assembly. The Trimmomatic16 with parameters (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50) was used to remove adapters and low-quality fragments of the raw Hi-C reads data. The processed reads were then aligned to the assembly using the Juicer (v1.6)17 with default settings. Contigs were scaffolded using 3D-DNA pipeline18 with all valid Hi-C reads. We use the Juicebox (v2.13.07)17 to adjust the chromosome-scale scaffolds manually(Fig. 2, Table 4). And there are 141 gaps among the 24 chromosomes.

Genome-wide Hi-C interaction mapping of chromosome sections.
Repeat annotation
We used de novo prediction and homology comparison to annotate the genomic repetitive sequences. RepeatModeler19 were used to detected and classified the repetitive sequences in the genome assembly using tools including RECON(v1.08)20, RepeatScout(v1.0.5)21, LTR-FINDER(v1.0.5)22 and TRF (v4.0.935)23. For homology comparison, RepeatMasker (open-4.0.9) and RepeatProteinMask (open-4.0.9) were used to identify the known TEs of the yellow-cheek carp genome in the Repbase TE library24,25 and TE protein database, respectively. The results showed that the genome repetitive sequence size was 456.66 Mb, accounting for 55.17% of the assembled genome. Among the repeat elements, short interspersed nuclear elements (SINEs) accounted for 0.24% of genome size and long interspersed nuclear elements (LINEs) accounted for 7.67%. Long terminal repeats (LTRs) and DNA elements accounted for 12.31% and 34.87%, respectively (Table 5).
Protein-coding gene prediction and annotation
In this research, the ab initio gene prediction, homology-based gene prediction and transcript prediction were used to predicted protein-coding genes of the yellow-cheek carp genome. Prior to gene prediction, the assembled yellow-cheek carp genome was hard and soft masked using RepeatMasker. The ab initio gene prediction was performed using Augustus (v3.3.1)26,27 and Genescan (v1.0)28. Models used for each gene predictor were trained from a set of high-quality proteins generated from the RNA-Seq data. For the homology-based prediction, Glimmer HMM(v3.0.4)29 was used to align the protein sequences to our genome assembly and predict coding genes with the default parameters. The reference protein sequences of five fish species, including Ctenopharyngodon idella, Sinocyclocheilus grahami, Megalobrama amblycephala, Danio rerio and Cyprinus carpio, were sourced from the NCBI database. For the transcript prediction, clean RNA-Seq reads were assembled into the yellow-cheek carp genome using Stringtie (v2.1.1)30. Then the gene structure was formed using PASA (v2.4.1)31. To consolidate the results from these three methods, MAKER (v3.00)32 was employed to enable the merging and integration of gene predictions.
For functional annotation of predicted gene, BLASTP (v2.6.0)33,34 was used to align the anticipated genes to the Kyoto Encyclopedia of Genes and Genomes (KEGG)35, Gene Ontology (GO)36, NCBI-NR (non-redundant protein database), Swiss-Prot37, TrEMBL38 and InterPro39 database. In total, we successfully predicted 24,153 protein-coding genes within the genome. These predicted genes displayed an average coding sequence length of 1638.21 bp, an average gene length of 18969.98 bp, and an average exon number of 9.87 (Table 6). Further, 22,965 genes, which accounts for 95.54% of the total number of predicted genes, were successfully assigned with at least one functional annotation (Table 7).
Annotation of non-coding RNA genes
The tRNAscan-SE (v1.3.1)40 algorithms with default parameters were used to identify the genes associated with tRNA. We downloaded the closely related species rRNA sequences from the Ensembl database. Then rRNAs in the database were aligned against our genome using BLASTn (v2.6.0)41 with E-value <1e-5, identity ≥85% and match length ≥50 bp. The miRNAs and snRNAs were identified by Infernal (v1.1.2)42 software against the Rfam (v14.1) database with default parameters. As a result, we annotated 76 rRNAs, 2469 tRNAs, 291 MiRNAs and 212 snRNAs (Table 8).
Data Records
All the raw sequencing data have been deposited in the NCBI database under the accession number SRP47030643. The genome assembly has been deposited at GenBank under the accession GCA_037101425.144. Genome annotations, along with predicted coding sequences and protein sequences, can be accessed through the Figshare45.
Technical Validation
The BUSCO was used to evaluate the quality of the genome assembly. We assessed assembly completeness using BUSCO (v3.0.259)46 with the reference arthropod gene set (n = 3,640). The final genome assembly showed a BUSCO completeness of 98.4%, consisting of 3,538 (97.2%) single-copy BUSCOs, 45 (1.2%) duplicated BUSCOs, 26 (0.7%) fragmented BUSCOs, and 31 (0.9%) missing BUSCOs (Table 9). Comparison of BUSCO results with Squaliobarbus curriculus (95.8%) and Mylopharyngodon piceus (96.0%) revealed the high genome assembly quality of yellow-cheeked carp47.
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software. No specific code has been developed for this study.
References
Zhu, N. S. & Chen, H. X. Food habits of yellow-cheek carp in Liangzi lake. Acta Hydrobiologica Sinica. 03, 262–271 (1959).
Liang, Z. S., Yi, B. L. & Yu, Z. T. Reproductive habits and embryonic development of yellow-cheek carp in the main stream of the Yangtze River and the Han River. Acta Hydrobiologica Sinica. 04, 389–403 (1984).
Ma, X. F., Wang, W. M. & Yang, Z. L. Biochemical composition and nutritional characteristics of yellow-cheek carp. Journal of Huazhong Agricultural University. 06, 759–762 (2008).
Yi, C. P. & Zhong, C. M. Yellow-cheek carp fat content determination and fatty acid composition analysis. Food Science. 14, 255–258 (2013).
Zhang, Z. Q. et al. Yellow-cheek carp meat rate and muscle nutrient analysis. Tianjin Agricultural Sciences. 04, 29–33 (2013).
Zhu, T. B. et al. Lushan west sea yellow-cheek carp national aquatic germplasm resources protection zone aquatic biological resources preliminary investigation. Biotic Resources. 02, 188–193 (2021).
Qi, X. R. Survey of fishery resources in the upper Han River. Journal of Fisheries Research. 01, 21–32 (2022).
Liao, F. et al. Complete mitochondrial genome of Elopichthys bambusa (Cypriniformes, Cyprinidae). Mitochondrial DNA. 27, 1387–1388 (2016).
Han, X. L. et al. The AFLP analysis of yellow-cheek carp group genetic diversity. Journal of Nanjing Normal University (Natural Science Edition). 01, 110–114 (2009).
Yang, W. & Fan, Q. X. The specialization breeding technology of yellow-cheek carp. Animals Breeding and Feed. 09, 32–33 (2011).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 13, 1050–1054 (2016).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience. 7, 1–6 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biology. 35, 62–67 (2013).
Xiao, C. L. et al. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat Methods. 14, 1072–1074 (2017).
Walker, B. J. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE. 9, e112963 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Flynn, J. M. et al. RepeatModeler 2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics. 21, i351–i358 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 27, 573–580 (1999).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110, 462–467 (2005).
Jurka, J. Repbase Update: a database and an electronic journal of repetitive elements. Trends in Genetics. 16, 418–420 (2000).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research. 33, W465–W467 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research. 34, W435–W439 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of Molecular Biology. 268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 20, 2878–2879 (2004).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33, 290–295 (2015).
Haas, B. J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research. 31, 5654–5666 (2003).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
AltschuP, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic Local Alignment Search Tool. Journal of molecular biology. 3, 403–410 (1990).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics. 10, 421 (2009).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research. 40, D109–D114 (2012).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 25, 25–29 (2000).
Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research. 31, 365–370 (2003).
Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research. 28, 45–48 (2000).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Research. 43, D213–D221 (2015).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research. 5, 955–964 (1997).
Altschul, S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 25, 3389–3402 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics. 25, 1335–1337 (2009).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP470306 (2023).
NCBI GenBank. https://identifiers.org/ncbi/insdc.gca:GCA_037101425.1 (2023).
Li, S. Chromosome-level genome assembly of the yellow-cheek carp Elopichthys bambusa. figshare https://doi.org/10.6084/m9.figshare.c.6931267.v1 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution. 38, 4647–4654 (2021).
Xu, M. R. X. et al. Maternal dominance contributes to subgenome differentiation in allopolyploid fishes. Nature Communication. 14, 8357 (2023).
Acknowledgements
This work was supported by the Key Research and Development Program of Hubei Province (2021BBA233 and 2023BBA001). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.L., S.W. and Z.G. conceived this study. S.L., S.Q., Z.S. and Y.H. collected the samples and performed the experiments; S.L. and X.X. performed the research and analyzed the data. S.L. drafted the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, S., Xiong, X., Qiu, S. et al. Chromosome-level genome assembly of the yellow-cheek carp Elopichthys bambusa. Sci Data 11, 426 (2024). https://doi.org/10.1038/s41597-024-03262-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03262-8
