
Abstract
Most research on trinucleotide repeats (TRs) focuses on human diseases, with few on the impact of TR expansions on plant gene expression. This work investigates TRs' effect on global gene expression in Psidium guajava L., a plant species with widespread distribution and significant relevance in the food, pharmacology, and economics sectors. We analyzed TR-containing coding sequences in 1,107 transcripts from 2,256 genes across root, shoot, young leaf, old leaf, and flower bud tissues of the Brazilian guava cultivars Cortibel RM and Paluma. Structural analysis revealed TR sequences with small repeat numbers (5–9) starting with cytosine or guanine or containing these bases. Functional annotation indicated TR-containing genes' involvement in cellular structures and processes (especially cell membranes and signal recognition), stress response, and resistance. Gene expression analysis showed significant variation, with a subset of highly expressed genes in both cultivars. Differential expression highlighted numerous down-regulated genes in Cortibel RM tissues, but not in Paluma, suggesting interplay between tissues and cultivars. Among 72 differentially expressed genes with TRs, 24 form miRNAs, 13 encode transcription factors, and 11 are associated with transposable elements. In addition, a set of 20 SSR-annotated, transcribed, and differentially expressed genes with TRs was selected as phenotypic markers for Psidium guajava and, potentially for closely related species as well.
Similar content being viewed by others

Introduction
Microsatellites are genomic, multiallelic, and codominant molecular markers widely distributed across both the genome of prokaryotes and the nuclear and organellar genome of eukaryotes, consisting of tandemly repeated motifs that range from mono to decanucleotide1,2,3,4,5. Hence being called “simple sequence repeats (SSRs)”, or, on occasion, “short tandem repeats (STRs)”4. These SSRs are flanked by highly conserved regions, despite their own being hypervariable, due to their inherent high mutation rate of 10–2–10–6 events per locus per generation1,2,3,4,5. It is these particulars that make them suitable for genetic profiling6, parentage analysis, gene and molecular tagging, linkage mapping, molecular diagnostics, comparative and functional genomics, taxonomic unit identification, analysis of genetic diversity2,7,8, and improvement of breeding programs through germplasm characterization9.
Polymorphism at SSR loci arises from the decrease or increase in repeat number (and as it increases, so does the mutation rate)2. There are four mutation dynamics responsible for this variability: replication slippage, unequal crossing over, gene conversion, and retrotransposition7. The first two are a consequence of the repetitive sequence’s very tendency to form secondary structures, such as hairpin and triplex, on the DNA strand3,7,10,11. During replication, DNA polymerase slippage at hairpins may lead to mispairing bases that escape proofreading and the mismatch repair system, ultimately resulting in the occurrence of deletions or insertions in the repetitive motifs3,10. On the other hand, recombination may also culminate in modifications at SSR sites, through unequal crossing over or unequal sister chromatid exchange2,3,6,7. As hairpins form over synapsis, one of the homologous chromosomes receives a larger fragment, containing more SSR repeats than the other7. As for gene conversion, it is presumed that the substitution of one nucleotide for another (following the model of non-reciprocal exchange) can promote variation in SSR sequences, the same as with minisatellites7. At last, retrotransposition may also mediate SSR variability, though its mechanisms have yet to be elucidated7.
The number of microsatellites detected in any given organism will depend on the size of its genome. For several different plants, SSR abundance will increase along with genome size, whereas its density will decrease5. Ultimately, though, at least one SSR array will be found for every 10 Kb of eukaryotic DNA sequence12. But while there is no consensus on a universally most common motif size, triplets (or its multiples) have been well reported as the most frequent SSR class within coding sequences (CDS) in several plant clades, like eudicots, monocots, and less derived plants, so they’re the most likely to turn up, at least in these groups3,5,13,14,15.
The prevalence of trinucleotides in CDS stems from negative selection against frameshift mutations in these regions, from mutation pressure, and positive selection for single amino acid stretch2,3,15. Despite that, shorter trinucleotide arrays are favored over longer ones, seeing that the latter may destabilize during meiosis or gametogenesis, and so, seem to experience stronger selection, especially in places where the recombination rate is high3. Hence, regardless of their location within coding or non-coding regions, certain tri-arrays exhibit an inherent inability to be conserved for long periods3.
Although SSRs in general have once been regarded as neutral markers, it is well known now that they do affect gene expression, be it by epigenetic silencing, by modulating RNA structure and function, or by directly altering the protein sequence3,16,17,18. Either way, they end up assuming functional roles in molecular, cellular, and metabolic processes1,3,6,7,9,16,18,19. In fact, several human loci contain trinucleotide repeats, the length of which naturally falls into a typical range17,20. But, as it happens in more than forty human neurological disorders21, the trimer repetition expands and surpasses the standard motif region length, leading to a pathological state17,20,21. Such is the case for fragile X disorders, myotonic dystrophies, and neurodegenerative and polyglutamine diseases 17,21. For example, in the most common hereditary genetic ataxia, Friedreich's Ataxia, the untranslated GAA motif repeats more than 66 times, reaching up to 1300 repetitions, when it would normally be repeated 5 to 34 times6,10,16,17,20,21,22.
Thus, most scientific investigations focusing on trinucleotide repeat (TR) expansions primarily concerns human diseases. As far as we know, only a few studies address the effect of TR on plant gene expression22,23,24. Thanks to these studies, it is known that the GAA motif, which is normally repeated around 0 to 36 times in the global sampling of the Arabidopsis thaliana L. population, is repeated more than 400 times in the third intron of this species’ Bur-0 IIL1 (isopropyl malate isomerase large sub unit1) gene23,24. Homozygous individuals under heat stress (above 27 ºC) or UV-B exposure undergo a growth defect that results in irregularly impaired leaves, the iil phenotype22,23,24, implying that environmental conditions do affect gene expression in introns. Carriers of this deleterious genotype do not even progress to the flowering stage23. This effect stems from the biogenesis and local accumulation of siRNA, which epigenetically modify the IIL1 locus through RNA-dependent methylation, down-regulating it10. However, one could argue that this same genotype — deleterious within the natural range of A. thaliana—could be advantageous, for example, in the Burren environment, where individuals would benefit from delayed flowering on long days, seeing as they would avoid late frosts; posing, therefore, a matter of antagonistic pleiotropy24. And indeed, SSRs are related to cryptic genetic variation as well as recent evolution24,25. Tabib et al. (2016) even showed how GAA expansion in wild populations has persisted for at least 60 years. Genic regions can therefore be used as microsatellite markers in population studies related to phenotypic variation.
As no previous study has approached yet the effect of TRs in plant CDS, Psidium guajava L. (family Myrtaceae) was chosen to further research on this subject. P. guajava (2n = 2x = 22)26 is a perennial, fruit tree that originated in the American tropic27,28,29, from where it dispersed, reaching wide into the world27. Now it is found largely in the tropics and subtropics, where its fruit—guava—is produced mainly by countries like India, China, Indonesia, Thailand, Pakistan, Mexico, and Brazil, respectively27,30. Besides its fruit, each and every part of its anatomy is exploitable in favor of its countless active principles. For example, its roots, stem, bark, and leaves are also rich in antiseptic, anti-bacterial, anti-inflammatory, antihypertension, diuretic, astringent, antispasmodic, cough sedative, antidiarrheic, anti-obesity, antitumor, anticancer, and cytotoxic proprieties, thus being capable of treating maladies of the gastrointestinal and respiratory tracts, skin, and wounds. Other compounds also treat diabetes, rheumatism, and general9,31,32,33,34,35. This vast herbal application is based on the myriad of secondary metabolites produced by this species31,33,34,35, which evidence its adaptation to disparate biogeographic conditions. On the other hand, its leaf isolates serve as the reducing agent for the green synthesis of iron magnetic nanoparticles36, which are of ample value to nanobiotechnology, being lately employed in oil spill remediation37, biodiesel synthesis38, protein loading39, as well as detection, imaging, and targeting of tumors40.
So, this work aims to investigate the effects of trimer repeats on the global gene expression of P. guajava, focusing on two genetically divergent Brazilian cultivars, namely Cortibel RM and Paluma. The Paluma cultivar is the most widespread in Brazil, characterized by a robust genotype, with easy propagation41. Concurrently, the cultivation of Cortibel guava has increased in the Brazilian Southwest because of its high productivity, and extended shelf life, making them suitable for export42.
Transcript levels and differentially expressed genes containing trimer repeats were examined across five distinct tissues (roots, shoots, young leaves, old leaves, and flower buds) in the Paluma and Cortibel RM using Illumina RNA-Seq. Trimer repeats were characterized considering both their size and the Gene Ontology annotations associated with their genomic loci. Among these predicted genes, those exhibiting greater stability and variability in terms of gene expression were selected to develop trinucleotide microsatellite markers involved with potential phenotypic changes. The resultant markers will subsequently be employed to validate the observed differential gene expression patterns.
Methods
Plant material, rna extraction, transcriptome assembly and alignment
The seedlings grew in nurseries. Tissues from two Brazilian cultivars, Cortibel RM and Paluma, comprising root, shoot, young leaf, and old leaf, were collected after a growth period of one hundred days. At the time of sampling, only Cortibel RM exhibited the presence of flower bud tissue. Consequently, this specific tissue was not obtained from Paluma. The twelve most uniform plants of each cultivar were selected by their morphological characteristics, as to ensure sample homogeneity. Four samples were collected from each of the above-mentioned tissues and mixed in equal proportion to form two pools of each cultivar. The total RNA was extracted using the CTAB method, chosen for its efficiency in yielding high-quality RNA across various genotypes, tissues, and stress conditions of P. guajava43.
The quality of total extracted RNA was measured with TapeStation System (Agilent), and the quantity with Qubit (Thermo Fisher Scientific). Samples with an RNA Integrity Number (RIN) greater than 5 were considered to be of high quality44.
The high-quality RNA was further subjected to rRNA removal using the RiboMinus Plant kit (Thermo Fisher Scientific). Libraries were prepared using Illumina TruSeq RNA Library Prep Kit, following the manufacturer's guide. Quantification was done with Qubit (Thermo Fisher Scientific), and fragment sizes were estimated using the TapeStation System (Agilent).
We sequenced two libraries from each tissue with Illumina Nextseq500 in Midi mode, generating paired-end reads of 75 bp. In the end, there were 18 libraries. The Illumina FASTQ files were analyzed with the NGS QC Toolkit software45 to check the quality of the sequenced reads, as well as the presence of artifacts that stem from the preparation of the sequenced samples, such as adapters and low-quality sequences, or from the sequencing itself.
The sequences were assembled de novo with Trinity Galaxy (version 2.15.1)46,47, where the sequences were grouped into contigs and singlets. The annotation was done by comparison with plant amino acid sequence databases. The contigs were also compared using the curated RefSeq48, Swiss-Prot49, TrEMBL49, and NCBI amino acid sequence50 databases. We processed the assembly and annotation process with the Galaxy instance of the Autralia platform (https://usegalaxy.org.au/). The best resulting alignment was selected to annotate the putative transcripts.
The eighteen RNA-Seq libraries were filtered, then mapped with BWA-MEM (Galaxy version 2.2.1) to the P. guajava’s draft genome, which is under submission. This alignment's read counts were used in PCA, relative abundance, and differential expression analyses.
Expression profile analysis of the RNA-seq data
Differential expression analysis requires the preprocessing of RNA-Seq data through filtering and alignment, for this purpose we used DESeq2::deseq()57 within R (Core Team, 2022). This methodology is based on the negative binomial distribution, applied to calculate the variation in transcript expression among different samples, as well as its statistical significance. Thus, the comparisons were made at the tissue level, between cultivars, arbitrarily choosing to use Cortibel RM as control and Paluma as treatment. Only genes present in both biological repeats were accepted, as to ensure data processing quality. Genes were considered differentially expressed when one duplicate of a cultivar displayed a significantly different mean from the other by the Student’s t-test (p-value < 0.05), being two times higher or lower (fold-change) than the other. We applied the FDR (False Discovery Rate) statistical test58 on the statistical significance of gene expression values between samples to filter out false positives.
Read counts underwent Z normalization, and the new Z values served as input to Principal Component Analysis (PCA), using R software v.0.55 (Core Team, 2022) with the packages::functions stats::prcomp59 and plotted using ggbiplot::ggbiplot60. A Ward-MLM (Modified Location Model) and UPGMA (Unweighted Pair Group Method with Arithmetic Mean) clusters were also generated using gplots::heatmap.261 for the relative abundance analysis while we used the package::heatmaply::heatmaply() function85 for the differential expression profiles.
Mining of SSRS, functional annotation, and enrichment analysis
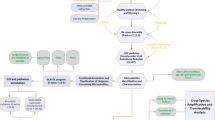
The genome, genes, and CDS were mined for SSR with MISA5,62. The parameters were set to default as follows: monomers ≥ 10, dimers ≥ 6, while trimers, tetramers, pentamers, and hexamers ≥ 5. A Python script assigned gene ontology identifiers to the genes containing trimer repeats, based on results from the InterPro63 and eggNOG64 databases. We retrieved the GO terms and their enrichment analysis with GOSlimViewer65 and KOBAS-i66, using this tool's default parameters. All charts except for those of enrichment were built in R Studio v 4.2.1. The flow chart in Fig. 1 depicts the summary of our methodology.

Overview of the methodology implemented in this study. Initially, we employed de novo assembly techniques to construct a hybrid draft genome using the Cortibel RM and Paluma cultivars. Subsequently, the MISA tool was utilized to identify microsatellites within the genome, gene, and coding sequences, resulting in the detection of 69,991 SSR, 33,978 SSR, and 4576 SSR, respectively. Functional annotation of the coding sequences was performed using Gene Ontology and InterPro Scan. Additionally, an enrichment analysis was conducted using KOBAS-i to determine the functional profile of the coding sequences. RNA-Seq data (read counts) of the CDS with TR from the root, shoot, young leaf, old leaf, and flower bud of both cultivars’ tissues served as input to PCA, relative abundance, and differential expression analysis. We identified microRNA (miRNA), transcription factors (TFs), and transposable elements (TEs) in genes with triplet repeats on the miRBase, Plant TF DB, and DPTE databases, respectively. Primers were then designed for the differentially expressed and upregulated genes that had functional annotation, but also for those in which the motif repeat number was unusually high (equal to or above 11), or for those in which the expression level was significantly high (equal to or above 5) in all tissues at the same time.
Criteria for putative gene selection and primer design
Among the entirety dataset of genes containing triplet repeats, a filtering process was employed, focusing on those demonstrating transcription with Z-values (normalized read counts) equal to or greater 1 in any tissue of both Cortibel RM and Paluma. Subsequently, the selection was refined to encompass differentially expressed genes that exhibited upregulation and had been annotated by either Gene Ontology (GO) 67,68 or InterProScan69. We also prioritized genes possessing an unusually high Z value (z > = 5) in all tissues at once, as well as those in which the motif’s repeat number was notably large (RN > 11).
Primers for coding sequences with TR were designed in NCBI PrimerBLAST70, avoiding annealing at exon-junction regions. The design specifications were: guanidine-cytosine content of 50–60%, melting temperature (MT) of 60–62 ºC, primer size of 20 to 22 nucleotides, and amplicon length of 110 to 180 base pairs. Hairpins and dimers were evaluated by IDT OligoAnalyzer71. Primers with ΔG values between -3 kcal/mol and -9 kcal/mol were considered the best primers.
The genes were amplified by PCR in 20 μL reactions using GoTaq® DNA.
Polymerase (Promega, USA), following the manufacturer's instructions.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Results and discussion
We opted to mine the genome, genes, and coding sequences for motifs consisting of monomers up to hexamers. The result is shown in Fig. 2A, where the two most represented classes of SSRs — dimers and trimers—are evident (Supplementary Table S1). Our results corroborate with previous descriptions3, in which dimers and their multiples are identified as the most frequent in non-coding sequences, and trimers and their multiples as the most frequent in CDS. Here, however, only dimers (but not their multiples) have high frequencies in genic and intergenic regions, and only trimmers (but not their multiples) display high frequencies in CDS. Since this work aims to develop putative markers for phenotypic variation, we stuck to the coding sequences and specifically, we concentrated our efforts on exploring the trinucleotide repeats within the coding sequences, as this SSR class represents the most abundant category.

Structural annotation of the SSRs detected by MISA within the Psidium guajava genome. (A) Amount of SSRs detected in the genome, genes, and coding sequences of Psidium guajava, arranged by motif size. A total of 69,991 SSRs were identified in the genome (dark blue), 33,978 SSR in genes (light blue), and 4.576 SSRs in CDS (yellow). Out of the 7572 trinucleotides found in genic regions, 3084 occur in CDS (i.e., exons). (B) Region and number of trinucleotides within 6213 genes (a total of 7572 trinucleotides were detected). The proportion of trimers found in each genetic region is calculated by dividing the number of trimers found in each region by the total number of trimers detected (e.g.: 3730 ÷ 7572 = 49.23%).
The Venn Diagram (Fig. 2B) demonstrates that a higher proportion of triplets are exclusively observed within intronic regions (49.23%) compared to any other genomic region. Exons have the second highest occurrence (31.65%) of trinucleotides in gene regions, followed by the 5'-UTR promoter region (12.8%). A small percentage of trinucleotides occur in 3'-UTR regions (4.39%), and an even smaller percentage in intron–exon junctions (4.1%). No trinucleotide occurs only upstream or downstream of introns and exons.
Although at first glance Fig. 2B does not seem to support the results found in Fig. 2A, since there seem to be more triplets in introns than in exons, one should consider the longer length or higher frequency of introns than exons in a genome72. When evaluating the intron/exon ratio, the larger density of trimers in exons stands out, sometimes containing more than a single motif within a sequence. As mentioned earlier, the frequency of triplets (and their multiples) is higher regarding other SSR classes in CDS due to selective pressure against frameshift or the stretching of some specific amino acid in the protein.
The smallest repeat numbers and motif lengths of triplet microsatellites are the most common (Fig. 3A; Supplementary Table S2). Especially in exons, since intronic sequences are more likely to carry longer repeats than CDS (Supplementary Table S1, S3). In A. thaliana, less than 1% of triplets exceed 5 repeats, with no expressed gene containing more than 41 repeats23. Also, the Bur-0 genotype, which features trinucleotide expansion, is rare among A. thaliana strains23. Low repeat numbers are more common, probably because short microsatellites are the starting point for subsequent extension of their length5. They are also more stable than long SSRs3, which are more prone to mutation, gaining repeats faster than shorter arrays. Natural selection also acts against repetitive sequence growth2,3,73, resulting in long SSR sequences also being more prone to losing repeats than shorter SSRs2.

Structural annotation of trimer motifs in guava CDS. (A) Absolute frequencies of the number of repeats (left) and trimer motif region lengths in CDS. (B) Occurrence number of the most represented trimer motifs in CDS.
In Fig. 3B, we see the top ten occurring motifs in CDS (Supplementary Table S3, S4). Among them, the five most frequent ones (AGG, AAG, CCG, AGC, and ACC, respectively) have at least one guanine or cytosine. GC content is always more enriched in genic regions, and indeed, GC content in this draft genome was 39.49%, while in CDS it was 49.68%—a measure similar to that of other eudicots. In addition, the frequency of these bases is closely related to epigenetic control via methylation74. Eimer et al.22 demonstrated in their experiment, where the accumulation of siRNAs and subsequent RNA-dependent methylation of the IIL1 locus caused the downregulation of this gene, setting off the iil phenotype. As shown later, several of the 72 differentially expressed genes from our gene set are downregulated in at least one tissue.
Moreover, the motifs above-mentioned encode amino acids that, when present in the primary structure of a protein, cause drastic changes in its native conformation. The AGG and AAG motifs, for example, encode positively charged amino acids (arginine and lysine, respectively) that result in electrical attraction or repulsion in the protein's three-dimensional structure75. This is worth noting because electrostatic interactions play multiple roles in protein-RNA complexes, such as the formation of the initial complex and its maintenance75,76. Arginine, in particular, is an effective suppressor of protein interactions77. The CCG motif, on the other hand, encodes proline, an amino acid that produces disruption in the secondary structure whenever present78. The AGC and ACC motifs encode serine and threonine, respectively—two polar, uncharged amino acids that can affect protein stability79.
When the genes harboring trimer repeats were transcribed, the expression values ended up centering mostly around motifs beginning with cytosine or guanine, with the motifs containing these bases displaying the highest means (Supplementary Figure S1-A). Likewise, the genes with the lowest repeat numbers (5–9) were the most expressed (Figure S1-B). On one hand, this would be expected from SSRs in coding sequences since these are conserved regions and the lower inherent mutation rate of shorter microsatellites means more stability in its region length. On the other hand, high level expression of a gene containing long SSR lengths is rare. The literature also points to highly repeated motifs being accompanied by low expression, as is the case of A. thaliana Bur-0 strain’s iil phenotype and Friedriech's ataxia22. Despite this, repeat numbers 16 and 17 display higher means than the smallest ones, where most of the expression data concentrated (Figure S1-B). In humans, large AC repeats in the NOS1 gene promoter lead to high expression levels, with shorter AC lengths being associated with impulsive behavior7. Long lengths of composed SSR sequences of poly-AC and poly-AG upstream of the transcription start site of the PAX6 human gene induce high expression levels, which is associated with myopia7. In our dataset, the 5'-UTR promoter region has the third-highest frequency of triplets in genes (Fig. 2B). Further exploration of this data would be required after Srivastava et al. (2019)15 pointed out that the preference for the accumulation of a particular class of long repeats may indicate selective pressure on these elements in an organism-specific manner.
If the largest part of this dataset refers to slightly repeating trimer SSR sequences, then Fig. 4 shows their gene ontology related to molecular function, cellular component and biological process. Cellular, intracellular, and cytoplasmic structures are not only the most abundant GO terms, but also the most represented, judging from their enrichment ratio in Fig. 5A and the relevance of the glycerolipid and glycerophospholipid metabolism clusters (the groups of overrepresented genes66) in Fig. 5B.

Gene Ontology functional annotation of the 1,107 transcribed genes containing trinucleotide.

Functional profile ofthe 1,107 transcribed genes carrying trimer repeats by KOBAS-ienrichment analysis, using Eucalyptus grandis genome as a reference (the closest relative of P. guajava84).
SSRs. A) Number of genes with trimer repeats associated with molecular functions. B) Number of genes with trimer repeats associated with cellular components. C) Number of genes with trimer repeats associated with biological processes.
A) Clusters of statistically significant functional terms associated with the transcripts containing trimer repeats and their enrichment ratio. B) Bubble plot of the scattering of p-values for each cluster and their functional terms. The node size, from small to large, represents the six levels of p-value (or R scores) — 0, 0.5, 1, 1.5, 2, 2.566.
The principal coordinates analysis (PCA) of transcribed genes carrying triplet SSRs in Supplementary Figure S2 shows the similarity between the experimental replicates, as well as the differences between the tissues in their expression profiles. These differences are further validated in the relative abundance analysis (Fig. 6A:C; Supplementary Table S5, S6). There is a large variation in gene expression ranging from -15 to 15 z-values, although most of the data centers around − 0.5 to 0.5 z-value (Fig. 7-A). A small set of genes is highly expressed, independent of the cultivar (Fig. 6A). Thus, the discrepancy in the read counts normalized to z-values is so high that the few upregulated genes end up masking the expression level of the others (Fig. 6A). These upregulated genes also appear to be highly expressed in all tissues (Fig. 6B), although one group of genes, in particular, is highly expressed only in Cortibel RM's flower bud and no other tissue. The chlorophyllated tissues usually cluster together, except when looking at Cortibel RM downregulated genes (Fig. 6C), whose effect can be seen in old leaves, where most genes are more expressed than other tissues. In addition, Cortibel RM downregulated root genes are clustered with the leaf tissue in Fig. 6C. These same effects are not observed in Paluma. Overall, we can see that the expression profiles and clusters are similar, but not identical. There is an effect of genes with trimer repeats between tissues and cultivars.

Ward-MLM and UPGMA hierarchical groupings of the normalized read counts of the 1,107 transcribed genes carrying triplet SSRs from the tissues root, shoot, young and old leaves, and flower bud of Cortibel RM and Paluma cultivars. (A) Relative abundance of all 1,107 transcribed genes with TRs. (B) Relative abundance of the 142 upregulated (z-score > 1) genes with TRs. (C) Relative abundance of the 811 downregulated (0,5 < z-score < 0) genes with TRs.

Differential expression of the 72 genes with trimer repeats from the roots, shoots, young and old leaves* of Cortibel RM (control) and Paluma (treatment) cultivars. (A) Differentially expressed genes harboring triplet SSRs from the roots, shoots, young and old leaves of both cultivars.The darker the blue, the more downregulated the gene is, and the redder the red, the more upregulated the gene is. (B) Amount of functionally annotated differentially expressed genes associated with transcription factors (TF), micro.
This effect can be further observed in Fig. 7A, the heatmap of differential expression between tissues of Paluma, as opposed to Cortibel RM. Genes such as Pg18407, Pg19499, Pg23742, Pg24087, Pg27787, and Pg34631 are much less expressed in Paluma than in Cortibel RM. These genes are related to sulfate transport and the EF-hand1 calcium-binding site, iron-sulfur cluster, UDP-Glycosyltransferase, sulfate transport and GTPase, and the B3 DNA-binding domain. Pg16449 is another gene that seems more downregulated in Paluma than in Cortibel RM, but it hasn’t been annotated either by GO or InterProScan. On the other hand, the genes Pg20686, Pg25676, Pg26960, and Pg36914 are much more expressed in Paluma than in Cortibel RM, being associated with RNA recognition and binding, dirigent proteins, aldehyde dehydrogenase and potassium ion transport, and cobalamin-independent methionine synthase, respectively. Though the Pg32594 gene is also much more upregulated in Paluma than in Cortibel RM, no annotation was found by GO or InterProScan.
Figure 7A also shows that shoots are the tissues with the highest number of differentially expressed genes containing trimer repeats, having the most variable expression levels as well. This was to be expected, considering that shoots are tissues undergoing differentiation. In contrast, we have the root as the tissue with the least number of differentially expressed genes but showing quite a difference in their expression. Young and old leaves have a close number of genes with differential expression.
The transcribed genes sum is 1,107 (from a total of 2,256 harboring TRs), of which 72 are differentially expressed (Suplementary Table 8). The Fig. 7-B summarizes the count of microRNAs, transcription factors, and transposable elements (Supplementary Table S7). Twenty-four were annotated by miRBase as stem-loop (also known as hairpin or hairpin loop) forming pre-miRNA — secondary structures on single strands of RNA formed by regions with complementary sequences when read from opposite sides —, very commonly found in microsatellite sites. Thirteen, annotated by the Plant TF database, encode transcription factors, involved with the ARF family proteins, bHLH, B3, DBB ERF, HD-ZIP, MYB, Trihelix, WRKY, prothodermal factor 2, ethylene-responsive element binding factor, telomere repeat binding factor 1, auxin response factor 9, B-box zinc finger protein with CCT domain, and with the NF-X-like 1, a gene whose product functions as a negative regulator of the defense response induced by trichothecene phytotoxins83. In fact, when analyzing the annotations of the entire gene set (2,256) as a whole, it is clear that many are resistance or stress response genes (which goes back to Fig. 4). Lastly, the remaining eleven differentially expressed genes were annotated by the Dioecious Plants Transposable Elements database (DPTEdb), with eight of them being helitron transposons and three being retrotransposons — two long terminal repeats (LTRs) and one long interspersed nuclear element (LINE).
RNAs (miRNA), and transposable elements (TE). *Since flower bud RNA was extracted for Cortibel RM alone, such tissue wasn't included in this analysis for comparison reasons.
In short, MISA identified 3,084 trinucleotides in 2,256 genes (Supplementary Table S8). Less than half are transcribed (1,107). But among them, only 72 are differentially expressed. Thirty-nine of these are upregulated, but only twenty-three have been annotated either by Gene Ontology or InterProScan. Some other genes with trimer repeats have large repeat numbers (NR; genes Pg11715, Pg32078, Pg47442, Pg47443, Pg50279) or very high normalized expression levels (EL > = 5) in all tissues simultaneously (genes Pg19110, Pg21266). So, primers were designed for 20 genes, of which the relation of gene ID, SSR motif, primers, and functional annotation is shown in Table 1. Finally, polymorphisms were identified in the Paluma and Cortibel RM gDNA, indicating putative functional markers for the species (Supplementary Figure S3). These results highlight the potential for development of functional SSR markers for P. guajava.
Conclusion
The SSR repeats in coding sequences potentially influence their activity and function, primarily attributed to variations in the repeat length causing phenotypic alterations in the plants. The trimer repeats was the predominant class observed within conserved coding sequences of Psidium guajava. Exploring the polymorphic set of markers, we characterized microsatellites with SRR trimer repeats, showing variation of expression across two important brazilian cultivars. We developed 20 markers related to primary metabolism (Pg16499, Pg19499, Pg34311, Pg35383, Pg37148, Pg37450) and secondary metabolism (Pg33230, Pg14098, Pg20103, Pg20401, Pg33733). This observation suggests the SSR expansion may impact processes related to the maintenance and adaptation of these cultivars, particularly on cellular structures and processes, such as cell membranes and signal recognition, or on stress responses and resistance. The development of markers targeting these specific sites associated with regulatory genes holds potential applications from molecular diagnostics to breeding programs. Also, SSR markers designed for Psidium guajava can be possibly utilized for other closely related species. The triplet SSR sites within these genes present opportunities for evolutionary studies and investigations into methylation process, considering both the repeat characteristics and their GC content.
Data availability
The supplementary material with the genes with TR studied were supplied. The transcriptomic data can be obtained through the specified accessions numbers, PRJNA1020439(https://www.ncbi.nlm.nih.gov/sra/PRJNA1020439).Thedatasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Experimental research and field studies on plants (either cultivated or wild), including the collection of plant material, must comply with relevant institutional, national, and international guidelines and legislation.
References
Buschiazzo, E. & Gemmell, N. J. The rise, fall and renaissance of microsatellites in eukaryotic genomes. BioEssays 28, 1040–1050 (2006).
Cavagnaro, P. F. et al. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 11, 569 (2010).
Li, Y.-C., Korol, A. B., Fahima, T., Beiles, A. & Nevo, E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review: Microsatellite evolution. Mol. Ecol. 11, 2453–2465 (2002).
Oliveira, E. J., Pádua, J. G., Zucchi, M. I., Vencovsky, R. & Vieira, M. L. C. Origin, evolution and genome distribution of microsatellites. Genet. Mol. Biol. 29, 294–307 (2006).
Zhu, L. et al. Short Tandem Repeats in plants: Genomic distribution and function prediction. Electron. J. Biotechnol. 50, 37–44 (2021).
Kolpakov, R. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 31, 3672–3678 (2003).
Gharesouran, J., Hosseinzadeh, H., Ghafouri-Fard, S., Taheri, M. & Rezazadeh, M. STRs: Ancient Architectures of the Genome beyond the Sequence. J. Mol. Neurosci. 71, 2441–2455 (2021).
Tuler, A. C. et al. SSR markers: a tool for species identification in Psidium (Myrtaceae). Mol. Biol. Rep. 42, 1501–1513 (2015).
Padmakar, B. et al. Development of SRAP and SSR marker-based genetic linkage maps of guava (Psidium guajava L). Sci. Hortic. 192, 158–165 (2015).
Lenzmeier, B. A. & Freudenreich, C. H. Trinucleotide repeat instability: a hairpin curve at the crossroads of replication, recombination, and repair. Cytogenet. Genome Res. 100, 7–24 (2003).
Viguera, E. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 20, 2587–2595 (2001).
Tautz, D. Hypervariability of simple sequences as a general source for polymorphic DNA markers. Nucleic Acids Res. 17, 6463–6471 (1989).
Furuno, M. et al. CDS Annotation in Full-Length cDNA Sequence. Genome Res. 13, 1478–1487 (2003).
Song, X. et al. Comprehensive analysis of SSRs and database construction using all complete gene-coding sequences in major horticultural and representative plants. Hortic. Res. 8, 122 (2021).
Srivastava, S., Avvaru, A. K., Sowpati, D. T. & Mishra, R. K. Patterns of microsatellite distribution across eukaryotic genomes. BMC Genom. 20, 153 (2019).
Kelkar, Y. D. et al. What Is a Microsatellite: A Computational and Experimental Definition Based upon Repeat Mutational Behavior at A/T and GT/AC Repeats. Genome Biol. Evol. 2, 620–635 (2010).
Hannan, A. J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 19, 286–298 (2018).
Kalia, R. K., Rai, M. K., Kalia, S., Singh, R. & Dhawan, A. K. Microsatellite markers: an overview of the recent progress in plants. Euphytica 177, 309–334 (2011).
Verstrepen, K. J., Jansen, A., Lewitter, F. & Fink, G. R. Intragenic tandem repeats generate functional variability. Nat. Genet. 37, 986–990 (2005).
Paulson, H. Repeat expansion diseases. Handbook Clin. Neurol. 147, 105–123 (2018).
Verma, A. K., Khan, E., Bhagwat, S. R. & Kumar, A. Exploring the Potential of Small Molecule-Based Therapeutic Approaches for Targeting Trinucleotide Repeat Disorders. Mol. Neurobiol. 57, 566–584 (2020).
Eimer, H. et al. RNA-dependent epigenetic silencing directs transcriptional downregulation caused by intronic repeat expansions. Cell 174, 1095–1105 (2018).
Sureshkumar, S. et al. A genetic defect caused by a triplet repeat expansion in Arabidopsis thaliana. Science 323, 1060–1063 (2009).
Tabib, A. et al. A Polynucleotide Repeat Expansion Causing Temperature-Sensitivity Persists in Wild Irish Accessions of Arabidopsis thaliana. Front. Plant Sci. https://doi.org/10.3389/fpls.2016.01311 (2016).
Paaby, A. B. & Rockman, M. V. Cryptic genetic variation: evolution’s hidden substrate. Nat. Rev. Genet. 15, 247–258 (2014).
Marques, A. et al. Refinement of the karyological aspects of Psidium guineense (Swartz, 1788): A comparison with Psidium guajava (Linnaeus, 1753). Comp. Cytogenet. 10, 117–128 (2016).
Pommer, C. V. & Murakami, K. R. N. Breeding Guava (Psidium guajava L). In Breeding Plantation Tree Crops: Tropical Species (eds Jain, S. M. & Priyadarshan, P. M.) (Springer, New York, 2009).
Arévalo-Marín, E. et al. The Taming of Psidium guajava: Natural and cultural history of a neotropical fruit. Front. Plant Sci. 12, 714763 (2021).
Proença, C. E. B. et al. Diversity, phylogeny and evolution of the rapidly evolving genus Psidium L (Myrtaceae, Myrteae). Ann. Bot. 129, 367–388 (2022).
Food and Agriculture Organization, FAOSTAT. Crops and livestock products: mangoes, guavas, and mangosteens. Available at: https://www.fao.org/faostat/en/#data/QCL/visualize (2023).
Díaz-de-Cerio, E. et al. Health effects of Psidium guajava L leaves: An overview of the last decade. Int. J. Mol. Sci. 18, 897 (2017).
Kherwar, D. et al. Microsatellite (SSR) marker assisted assessment of population structure and genetic diversity for morpho-physiological traits in guava (Psidium guajava L). J. Plant Biochem. Biotechnol. 27, 284–292 (2018).
Kumar, M. et al. Guava (Psidium guajava L) leaves: Nutritional composition phytochemical profile and health-promoting bioactivities. Foods 10, 752 (2021).
Naseer, S. et al. The phytochemistry and medicinal value of Psidium guajava (guava). Clin. Phytosci. 4, 32 (2018).
Qin, X.-J. et al. Meroterpenoids with Antitumor Activities from Guava ( Psidium guajava ). J. Agric. Food Chem. 65, 4993–4999 (2017).
Madubuonu, N. et al. Bio-inspired iron oxide nanoparticles using Psidium guajava aqueous extract for antibacterial activity. Appl. Phys. A 126, 72 (2020).
Qiao, K. et al. Application of magnetic adsorbents based on iron oxide nanoparticles for oil spill remediation: A review. J. Taiwan Inst. Chem. Eng. 97, 227–236 (2019).
Bilal, M., Zhao, Y., Rasheed, T. & Iqbal, H. M. N. Magnetic nanoparticles as versatile carriers for enzymes immobilization: A review. Int. J. Biol. Macromol. 120, 2530–2544 (2018).
Gaspar, A. S. et al. Biocompatible and high-magnetically responsive iron oxide nanoparticles for protein loading. J. Phys. Chem. Solids 134, 273–285 (2019).
Israel, L. L., Galstyan, A., Holler, E. & Ljubimova, J. Y. Magnetic iron oxide nanoparticles for imaging, targeting and treatment of primary and metastatic tumors of the brain. J. Controll. Release 320, 45–62 (2020).
Bezerra, I. L. et al. Physiological indices and growth of ‘Paluma’guava under saline water irrigation and nitrogen fertigation. Revista Caatinga 31, 808–816 (2018).
Gomes, F. R. et al. Correlations between physical and chemical characteristics of Cortibel guava (Psidium guajava L.) fruits grown in the Brazilian Cerrado. Revista de la Facultad de Ciencias Agrarias UNCuyo 1(10), 16 (2023).
de Carpinetti, P. A. et al. Efficient method for isolation of high-quality RNA from Psidium guajava L tissues. Plos ONE 16, e0255245 (2021).
C.V., S. B. & Gassmann, M. Assessing Integrity of Plant RNA with the Agilent 2100 Bioanalyzer System.
Patel, R. K. & Jain, M. NGS QC toolkit: A toolkit for quality control of next generation sequencing data. Plos one 7, e30619 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
The UniProt Consortium et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 (2023).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 50, D20–D26 (2022).
Feng, C. et al. A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol. J. 19, 717–730 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at http://arxiv.org/abs/1207.3907 (2012).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience https://doi.org/10.1093/gigascience/giab008 (2021).
Bolser, D. M. et al. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomic Data. In Plant Genomics Databases (ed. van Dijk, A. D. J.) (Springer, New York, 2017).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/ (2022).
Vu VQ (2011). _ggbiplot: A ggplot2 based biplot_. R package version 0.55, <http://github.com/vqv/ggbiplot> (2022).
Warnes, G. et al. _gplots: Various R Programming Tools for Plotting Data_. R package version 3.1.3, <https://CRAN.R-project.org/package=gplots> (2022).
Biswas, M. K. et al. Transcriptome wide SSR discovery cross-taxa transferability and development of marker database for studying genetic diversity population structure of Lilium species. Sci. Rep. 10, 18621 (2020).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 (2019).
Huerta-Cepas, J. et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 (2016).
McCarthy, F. M. et al. AgBase: A functional genomics resource for agriculture. BMC Genom. 7, 229 (2006).
Bu, D. et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 49, W317–W325 (2021).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
The Gene Ontology group et al. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Ye, J. et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13, 134 (2012).
Owczarzy, R. et al. IDT SciTools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 36, W163–W169 (2008).
COOPER, Geoffrey M.; HAUSMAN, R. E. The complexity of eukaryotic genomes. The Cell: A Molecular Approach, 2 (2000).
Astolfi, P., Bellizzi, D. & Sgaramella, V. Frequency and coverage of trinucleotide repeats in eukaryotes. Gene 317, 117–125 (2003).
Shenker, N. & Flanagan, J. M. Intragenic DNA methylation: Implications of this epigenetic mechanism for cancer research. Br. J. Cancer 106, 248–253 (2012).
Andersson, H., Bakker, E. & von Heijne, G. Different positively charged amino acids have similar effects on the topology of a polytopic transmembrane protein in Escherichia coli. J. Biol. Chem. 267, 1491–1495 (1992).
Law, M. J. The role of positively charged amino acids and electrostatic interactions in the complex of U1A protein and U1 hairpin II RNA. Nucleic Acids Res. 34, 275–285 (2006).
Arakawa, T. et al. Suppression of protein interactions by arginine: A proposed mechanism of the arginine effects. Biophys. Chem. 127, 1–8 (2007).
MacArthur, M. W. & Thornton, J. M. Influence of proline residues on protein conformation. J. Mol. Biol. 218, 397–412 (1991).
Green, S. M., Meeker, A. K. & Shortle, D. Contributions of the polar, uncharged amino acids to the stability of staphylococcal nuclease: Evidence for mutational effects on the free energy of the denatured state. Biochemistry 31, 5717–5728 (1992).
Canal, D. et al. Exploring the versatility of sesquiterpene biosynthesis in guava plants: A comparative genome-wide analysis of two cultivars. Scientific Reports 14, 574 (2024).
Gershenzon, J. & Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414 (2007).
Farag, N. F. et al. Characterization of essential oils from Myrtaceae species using ATR-IR vibrational spectroscopy coupled to chemometrics. Ind. Crops Prod. 124, 870–877 (2018).
Asano, T. et al. AtNFXL1, an Arabidopsis homologue of the human transcription factor NF-X1, functions as a negative regulator of the trichothecene phytotoxin-induced defense response: Trichothecene-inducible AtNFXL1 gene. Plant J. 53, 450–464 (2007).
Thakur, S. et al. Development of genome-wide functional markers using draft genome assembly of guava (Psidium guajava L.) cv. allahabad safeda to expedite molecula breeding. Front. Plant Sci. 12, 708332 (2021).
Galili, T. et al. Heatmaply: An R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 34, 1600–1602 (2018).
Funding
This work was supported by Fundação de Amparo à Pesquisa do Espírito Santo (FAPES, Vitória — ES, Brazil; grant 75516586/16), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília — DF, Brazil; Finance Code 001), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasília — DF, Brazil; grants 443801/2014–2 and 308828/2015–1).
Author information
Authors and Affiliations
Contributions
G.P.P.: Structural and functional annotation, functional enrichment, relative abundance analysis, transcriptome alignment, variant calling, writing, review, and editing. D.C.C.: Structural annotation. V.S.F.: primer design, validation, and writing. D.C.: Review and structural annotation. M.F.: Functional annotation. P.A.C.O.: RNA-seq experimental design and transcriptome obtention, hierarchical groupings and differential expression. O.J.B.B.: transcriptome alignment, assembly, annotation, and differential expression. A.F.: Conceptualization, funding acquisition, transcriptome assembly. M.F.S.F.: Conceptualization, supervision, review, and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pires, G.P., Fioresi, V.S., Canal, D. et al. Effects of trimer repeats on Psidium guajava L. gene expression and prospection of functional microsatellite markers. Sci Rep 14, 9811 (2024). https://doi.org/10.1038/s41598-024-60417-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-60417-8
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
