
Abstract
Yellow fever outbreaks are prevalent, particularly in endemic regions. Given the lack of an established treatment for this disease, significant attention has been directed toward managing this arbovirus. In response, we developed a multiepitope vaccine designed to elicit an immune response, utilizing advanced immunoinformatic and molecular modeling techniques. To achieve this, we predicted B- and T-cell epitopes using the sequences from all structural (E, prM, and C) and nonstructural proteins of 196 YFV strains. Through comprehensive analysis, we identified 10 cytotoxic T-lymphocyte (CTL) and 5T-helper (Th) epitopes that exhibited overlap with B-lymphocyte epitopes. These epitopes were further evaluated for their affinity to a wide range of human leukocyte antigen system alleles and were rigorously tested for antigenicity, immunogenicity, allergenicity, toxicity, and conservation. These epitopes were linked to an adjuvant (\(\beta\)-defensin) and to each other using ligands, resulting in a vaccine sequence with appropriate physicochemical properties. The 3D structure of this sequence was created, improved, and quality checked; then it was anchored to the Toll-like receptor. Molecular Dynamics and Quantum Mechanics/Molecular Mechanics simulations were employed to enhance the accuracy of docking calculations, with the QM portion of the simulations carried out utilizing the density functional theory formalism. Moreover, the inoculation model was able to provide an optimal codon sequence that was inserted into the pET-28a( +) vector for in silico cloning and could even stimulate highly relevant humoral and cellular immunological responses. Overall, these results suggest that the designed multi-epitope vaccine can serve as prophylaxis against the yellow fever virus.
Similar content being viewed by others
Introduction
The Yellow fever virus belongs to the Flavivirus genus, a part of the Flaviviridae family1. This arbovirus, also known as an arthropod-borne virus, features a single-stranded, unsegmented RNA genome with positive polarity. This genome encompasses a single reading frame, spanning a total of 10,233 nucleotides. Within this genetic sequence, it encodes three structural proteins (E, prM, and C) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). These protein-coding regions are separated by a short noncoding segment2. The structural proteins contribute to the virus's basic structure, playing a essential function in the human immune activities, while the non-structural proteins are responsible for regulatory activities and virus expression3.
The discovery of the origin of this virus was possible only through phylogenetic analysis studies showing that the noncoding regions of the virus strains originating in Africa are much more conserved than those of the strains originating in the Americas1,2. This indicates that the virus originated on the African continent and spread to the Americas. The places most affected by the virus are the tropical regions of South Africa and the Americas, where its vectors (Aedes spp and Haemagogus spp) are endemic. From 2019 to 2021, the World Health Organization (WHO) documented yellow fever outbreaks in sixteen African countries (including Chad, Cameroon, Central African Republic, Côte d’Ivoire, Democratic Republic of Congo, Ghana, Niger, Nigeria, Republic of Congo, Senegal, Guinea, Gabon, Togo, Ethiopia, South Sudan, and Uganda) and three countries in the Americas (Venezuela, French Guiana, and Brazil). These occurrences raised significant concerns about disease control7.
Transmission of the virus occurs exclusively through the bite of the transmitting mosquito, with no direct human-to-human transmission. There are two transmission cycles of the virus: the urban cycle, in which the Aedes aegypti mosquito is responsible for spreading the disease, and the sylvatic cycle, in which several species are involved in transmission, the Aedes mosquitoes in Africa and the Haemagogus and Sabethes mosquitoes in the Americas3.
The incubation period of the virus is short, usually 3–6 days, but may be as long as 10 days. Yellow fever occurs in asymptomatic, mild and moderate forms with a nonspecific fever pattern that may or may not be accompanied by jaundice. However, it can also manifest in the severe form, which has a high mortality rate, with affected individuals exhibiting jaundice, organ dysfunction, and even hemorrhage4. There is no particular therapy for yellow fever at now, and in milder cases, treatment is supportive of symptoms. Only in severe cases are patients admitted to hospitals for more specific treatment, which is only available in intensive care units5. For this reason, the forms of prevention are crucially important in combating this disease, the most important being the vaccine.
Currently available immunizations against yellow fever are attenuated viral vaccines (17DD and 17D-204), which have been shown to be very effective in forming an immune memory. Approximately 90% of vaccinated individuals develop antibodies to yellow fever in less than 1 month6. Adverse effects associated with this vaccine are rare in healthy individuals, but are common in children, the elderly over 60 years of age and in individuals with weakened immune systems or hypersensitivity to vaccine components. In these cases, vaccination is not recommended and can lead to local, neurological and even systemic adverse effects7,8,9,10,11,12. While the 17D-204 vaccine has maintained an outstanding safety track record, it's crucial to acknowledge that rare instances (approximately 1 in 250,000 cases) of severe adverse effects have been reported, and, regrettably, these cases can be fatal. These adverse outcomes, which result from the neuroinvasion of 17D-204, are categorized as vaccine-associated neurotropic diseases. They encompass conditions such as post-vaccinal encephalitis, Guillain-Barré syndrome, and autoimmune disorders affecting either the central or peripheral nervous system13.
Although the 17DD vaccine has traditionally been associated with a protective humoral immune response against virulent YFV strains11, it remains unclear whether the T-cell immune response is also significant. Furthermore, instances of vaccine-induced multisystemic illness have been reported in Brazil, the United States, Argentina, and Australia, with fatal outcomes in most cases6,14,15,16. Furthermore, it's worth noting that vaccine viscerotropic diseases have been documented, which are distinguished by systemic infections affecting multiple organ systems, including liver damage that closely resembles the effects of wild-type infections17,18,19. Considering the recurrence of the diseases, the lack of drugs to treat it, and the side effects that the attenuated virus vaccine causes in certain groups, the development of a subunit vaccine that contains only a portion of the infectious agent is of paramount importance to minimize harm.
In light of this, the present work proposed to use immunoinformatic in order to predict parts of yellow fever virus that are compatible with an acceptable number of HLA alleles, antigenic, non-allergenic, immunogenic, non-toxic, conserved, and have good population coverage for cytotoxic T lymphocytes (CTL), helper T lymphocytes (HTL), and B cells.
From these epitopes, a vaccine template was generated and tested for its receptor binding affinity (TLR-2) and ability to elicit an immune response in the body. Additionally, the study included an assessment of gene expression related to the vaccine in E. coli. Furthermore, in silico gene cloning was conducted using the pET-28a( +) vector. This is a consolidated approach that has been used in several studies with pathogenic microorganisms such as SARS-CoV-220, Streptococcus pneumoniae21, and Alkhurma hemorrhagic fever virus 22 as well as with other arboviruses such as dengue23, chikungunya24, and Mayaro virus25,26.
Building upon this foundation, the objective of this research was to develop a multi-epitope vaccine targeting the yellow fever virus. This vaccine was designed to incorporate epitopes characterized by high levels of antigenicity, immunogenicity, conservation, non-allergenicity, non-toxicity, and exceptional population coverage, all with the capacity to induce a robust immune response within the human body. To achieve this goal, an exhaustive examination of the structural (E, prM, and C) and nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) across 196 sequenced viral strains was conducted, coupled with the application of a rigorous molecular modeling approach.
Methods
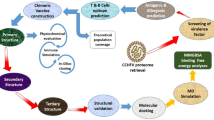
The flow chart of the methodology used in this study is shown graphically in Fig. 1. Recently, our research group validated similar immunoinformatics and molecular modeling approaches in the construction of a multiepitope vaccine against Mayaro virus and SARS-CoV-225,26,27.

A comprehensive schematic portrayal of the workflow employed in the present study.
Development/obtaining viral protein sequences
Prediction of T‑cell epitopes
Firstly, the primary sequences of the structural (C, M, and E) and nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins of the yellow fever virus (YFV) were obtained from the Virus Pathogen Resource (ViPR) database (https://www.viprbrc.org/brc/home.spg?decorator=vipr)28. The data were meticulously filtered according to specific criteria, including Family Flaviviridae, Genus Flavivirus, Species Yellow fever virus, and Global geographic group, leading to a preliminary selection of 385 sequences from the ViPR database; this was further refined to 196 sequences for detailed investigation based on additional criteria of ‘genome complete’ and ‘human host group’. This comprehensive analysis encompassed sequences from various geographical regions, including Africa (27), Asia (9), Europe (28), North America (7), and South America (125), thus ensuring a globally representative and diverse dataset for our yellow fever virus research. To achieve our research objectives, the proteomes of these 196 viral strains were meticulously compiled, categorized by protein type, and systematically aligned to derive consensus sequences.
We conducted MHC class I restricted (CTL) epitope prediction using two online servers: ProPred I (http://crdd.osdd.net/raghava/propred1) and NetCTL (http://www.cbs.dtu.dk/services/NetCTL/). ProPred I identifies promiscuous regions in protein sequences by employing matrices for 47 MHC-I alleles and models for proteasomal and immunoproteasome processing33. The analysis was carried out with specific parameters, including a 4% threshold, and the activation of proteasome and immunoproteasome filters, each set at a threshold of 5%, as referenced in34,35.
For validation of the identified epitopes in ProPred, we utilized NetCTL. NetCTL not only predicts potential epitopes within protein sequences for cytotoxic T lymphocytes (CTL) but also provides information regarding proteasomal C-terminal cleavage through artificial neural networks and TAP transport efficiency through weight matrix calculations36. The thresholds for CTL epitope identification, C-terminal cleavage, and TAP transport efficiency were set at 0.75, 0.15, and 0.05, respectively.
Similarly, two distinct methods were employed to assess the HTL epitopes capable of binding to HLA-DQ, HLA-DP, and HLA-DR alleles, employing artificial neural networks. The IEDB tool (http://tools.iedb.org/mhcii/) utilizes a big dataset including over 10,000 unpublished MHC-peptide binding affinities, 29 peptide/MHC crystal structures, and 664 peptides that have been experimentally tested29. To validate the forecasts generated by IEDB, the protein sequences have been uploaded to the NetMHCIIpan database., which is accessible at http://www.cbs.dtu.dk/services/NetMHCIIpan/. This server utilizes a vast dataset that includes more than 100,000 quantitative measurements of peptide binding sourced from IEDB. It covers a wide range of molecules, including HLA-DR, HLA-DQ, HLA-DP and even mouse MHC-II molecules, namely 36 HLA-DR, 27 HLA-DQ, 9 HLA-DP and 8 mouse MHC-II molecules38.
Prediction of B‑cell epitopes
The “Bepipred Linear Epitope Prediction 2.0” method, available through the IEDB tool at http://tools.iedb.org/bcell/, was employed to enhance the accuracy of predicting B lymphocyte (BL) epitopes within protein sequences. "Prediction 2.0" was specifically developed to address limitations observed in other epitope prediction tools, which predict continuous epitopes through a random forest algorithm trained on epitopes annotated from antibody antigen protein structures30. The standard threshold of 0.5 for predictions was maintained.
Antigenicity prediction
The theoretical epitopes underwent an antigenicity evaluation step in which they were individually entered into VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). This software assesses antigenicity, taking into account the selection of a target organism, such as a virus, bacterium, tumor, parasite, or fungus. In this analysis, a threshold of 0.5 was adopted, as this value aligns with the peak accuracy for most of the models used31.
Allergenicity prediction
The allergic or nonallergic nature of possible epitopes was predicted using the AllerTop 2.0 server (http://www.ddg-pharmfac.net/AllerTOP/). This online server evaluates the similarities between the peptide sequence under study and the sequences in its database. The epitopes are individually assessed, and the outcome, indicating whether the sequence is likely allergenic or non-allergenic, is furnished. Additionally, a link to the protein with a similar sequence is provided.
Immunogenicity prediction
Immunogenicity scores of CTL epitopes were calculated using IEDB immunogenicity (http://tools.iedb.org/ immunogenicity/). This tool collects the most important variables affecting immunogenicity, such as the P4-6 position of a peptide and amino acids with large and aromatic side chains32. The masking option used was the default (1, 2, and c-terminal) and the cutoff was set to zero33.
Toxicity prediction
To guarantee that the chosen epitopes were not toxic, the ToxinPred web server (http://www.imtech.res.in/raghava/toxinpred/) was used. To predict the toxicity of the epitopes, the server is based on the physicochemical characteristics of the peptides using machine learning and a quantitative matrix. The database of this method includes 1,805 toxic and 3,593 non-toxic peptides34.
Conservation analysis
To gauge the extent of epitope conservation within the acquired protein sequences at varying levels of identity, the IEDB conservation tool, accessible at http://tools.iedb.org/conservancy44, was utilized. The extent of conservation is measured by the percentage of protein sequences with which the epitope is identical at a given level of similarity. This approach allows for the selection of broadly protective epitopes.
Population coverage analysis
Population coverage provides a direct indication of vaccine efficacy in different geographic regions by examining the prevalence of human leukocyte antigen (HLA) alleles associated with the epitope of interest. For this study, the selected epitopes, along with their respective HLA-binding alleles, were submitted to the IEDB population coverage tool (http://tools.iedb.org/conservancy)35. This tool was programmed for the principal endemic regions of yellow fever—South America and Africa.
Multi–epitope vaccine construction
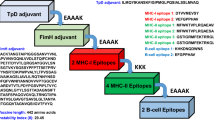
The vaccine sequence was constructed using LCTL and HTL epitopes with sequences overlapping with the BL epitopes that passed all immunoinformatic analyses. These sequences were connected using AAY and GPGPG linkers, respectively36. The AAY peptide linker helps the epitopes generate suitable sites for binding to the TAP transporter and enhances epitope presentation, whereas the GPGPG linker stimulates TCD4 + responses and preserves the conformational immunogenicity of the helpers as well as the antibody epitopes37.
A \(\beta\)-defensin adjuvant sequence was added to the N-terminus of the multi-epitope vaccine using the linker EAAAK, thus enhancing its immunogenicity. The \(\beta\)-defensin induces recruitment of naive T cells and immature dendritic cells by contacting TLR and CCR 6 (chemokine receptor 6) receptors at the site of infection38, and the linker EAAAK reduces association with other protein domains with efficient detachment and increases stability39.
Assessment physicochemical properties of the vaccine prototype
To analyze the physicochemical characteristics of the vaccine model, such as: To obtain information on molecular weight, pI (isoelectric point), half-life, instability index, aliphatic index, and GRAVY (Grand Average Hydropathy) of the vaccine sequence, it was subjected to analysis using the ExPASy-ProtParam tool, available at http://web.expasy.org/protparam/. These analyses leveraged a database of proteins with well-established properties, which were used as reference parameters for the provided protein sequence40.
Design, refinement and validation of the tertiary structure of the vaccine prototype
The Raptor-X server, accessible at http://raptorx.uchicago.edu/, was used to predict the three-dimensional structure of the vaccine sequence. This procedure consists of taking an input sequence in FASTA format and implementing three techniques: single and multiple template threading, along with prediction of alignment quality7. To gauge the precision of the projected 3D structure, the online website is the most trusted sources, which encompass the P-value for relative global quality, as well as GDT (Global Distance Test) and uGDT (un-normalized GDT) for assessing the overall structural integrity52.
The refinement of the tertiary structure was carried out through the GalaxyRefine 2 server, which is available at http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE2. This web server employs a specialized approach for refining 3D structures by implementing short Molecular Dynamics simulations after repeated perturbations involving side-chain repacking at both global and local levels. This method enables more extensive structural adjustments53. It incorporates multiple local and global move sets and iteratively accumulates conformational changes, facilitating larger-scale modifications. The local and global move sets use an estimated structure error to concentrate refinement efforts in regions with greater inaccuracies.
Finally, the quality and potential errors in the 3D model were checked using MolProbity, Swiss Model, ProSA-web, ERRAT, and Verify3D. For structural validation, the Swiss Model's Structure Assessment Tool, available at https://swissmodel.expasy.org/assess, was employed to obtain information on both the global and local aspects of the structure. This tool utilizes its own methods, including QMEAN and Ramachandran plot analysis, and can also run additional software tools like MolProbity. In addition, the ProSA web server, accessible at https://prosa.services.come.sbg.ac.at/prosa.php, was included in the analysis to validate the structural quality, and a quality score (Z-score) for the input structure was calculated. Score values that fall outside a typical range for native proteins suggest that there are likely errors in the structure41. Another validation server, known as ERRAT (http://services.mbi.ucla.edu/ERRAT/)42, assessed disjoint interactions within the framework. The accuracy of the 3D model's design was assessed using Verify3D, available at https://servicesn.mbi.ucla.edu/Verify3D/. This tool gauges the compatibility of the model with its corresponding amino acid sequence, providing insights into the quality and reliability of the structural model43.
Molecular docking simulations and refinement
Binding of the vaccine to the appropriate immunological recipient is paramount to elicit an appropriate immune response44,45. Toll-like receptors (TLR) are members of a family of pattern recognition receptors that recognize products of various microorganisms46. In the recognition of YFV by the host immune system, TLR-2 along with three other Toll-like receptors (7, 8, and 9) are described to be crucial for the interactions between 17D vaccine and human cells stimulating a mixed Th2 and Th1 cell profile47.
Therefore, the vaccinal model was linked to the TLR-2 receptor (PDB ID: 2z7x) using PatchDock server (http://bioinfo3d.cs.tau.ac.il/PatchDock/). PatchDock48 is a geometry-based molecular docking algorithm. This program, upon inputting two molecules, segments them into patches based on their surface shape, effectively dividing them into patterns akin to visually distinguishable puzzle pieces. The algorithm entails several key steps: (a) the representation of molecular shape, (b) matching surface patches, (c) filtering and evaluation. These patches can be overlaid using shape matching algorithms to facilitate the comparison and analysis of the two molecular structures49.
The FireDock web tool (http://bioinfo3d.cs.tau.ac.il/FireDock/)50 was used to optimize and re-evaluate the rigid-body molecular docking solutions. The final 10 models are categorized based on a general energy that includes atomic contact energy as well as van der Waals interaction, partial electrostatics, and binding energy estimates. The most promising Firedock model underwent further refinement within the HADDOCK interface, which can be accessed at https://bianca.science.uu.nl/haddock2.4/refinement/1. This server offers a list of clusters ranked by score and provides comprehensive statistics, including the average score for the top four structures within each cluster. This step allows for a more in-depth analysis and selection of refined structures based on their quality and score51.
Molecular dynamics simulation (MD) is an important technique for analyzing the strength of the receptor-ligand complex. It was used with the WEBGRO for Macromolecular Simulations (https://simlab.uams.edu/) to investigate the binding stability of the final complex52. In the simulation of the TLR2-vaccine complex, a 50 ns timeframe was employed. The GROMOS96 43a1 force field parameters were used for the simulation. The entire system was solvated in water, neutralized to balance the charges, and supplemented with 0.15 M NaCl salt to mimic physiological conditions. Key simulation parameters monitored during this process included the Root Mean Square Deviation (RMSD) and the Root Mean Square Fluctuation (RMSF). These parameters are fundamental for assessing the stability and dynamics of the system throughout the simulation.
To identify the most pertinent vaccine-TLR2 complex resulting from the docking calculations, we employed the combined quantum mechanics/molecular mechanics technique (QM/MM). This approach combines quantum mechanics for the ligand-receptor interactions with molecular mechanics for the surrounding environment, allowing for a more accurate representation of the system's behavior. QM/MM methods have solidified their position as advanced computational tools for studying biomolecular systems, as evidenced by the increasing number of publications utilizing these methods20,26,53,54. this procedure consists of taking an input sequence in FASTA format and implementing three techniques: single and multiple template threading, along with prediction of alignment quality7. To gauge the precision of the projected 3D structure, the server offers confidence scores, which encompass the P-value for relative global quality, as well as GDT (Global Distance Test) and uGDT (un-normalized GDT) for assessing the overall structural integrity52. The ONIOM multilayer technique, a unified strategy accessible in the Gaussian code, was used to carry out the QM/MM optimization. Ab initio calculations of the total energy of large complexes, such as biological systems, are possible and accurate using this approach when the systems have been divided into two or three layers. The TLR-2 receptor was placed in the MM layer, while the vaccine's main residues of amino acids were assigned to the QM layer.
To enlarge the electronic orbitals in the QM layer, we used the well-known B3LYP hybrid functional (Becke, three parameters, Lee–Yang–Parr) for exchange–correlation in conjunction with the 6-311G (d, p) basis set. Notably, during the geometry optimization process, all amino acid residues within a 6.0 Å radius from the ligand's centroid were allowed to adjust their positions55,56,57. This approach facilitates the accurate exploration of the ligand-receptor interactions and structural changes within the specified region of the complex 58.
The best PatchDock + FireDock + HADDOCK (PFH) and PatchDock + FireDock + HADDOCK + MD + QM/MM (PFHMQM) models were operated in the PRODIGY prediction for a comparative analysis of binding energies. The PRODIGY forecast protein–protein binding affinity (or binding free energy) on the basis of the biological system's structure and function, i.e., the interfacial contact network59. RMSD analysis in Discovery studio compared structures to original PatchDock complexes, revealing structural disparities60.
Finally, Discovery Studio Visualizer, LigPlot + (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/), and Pose View (https://proteins.Plus/) were implemented to evaluate binding postures and the existence of intermolecular interactions, in particular intermolecular hydrogen bonds (Fig. 2a) (carbon, conventional, and pi-donor H-bonds), electrostatic (Fig. 2b) (salt bridge, attractive charges, pi-cation, pi-anion), hydrophobic (Fig. 2c) (pi-pi stacked, pi-pi stacked, alkyl, pi-sigma, pi-alkyl), halogens (Cl, fluorine, Br, and I), miscellaneous (charge repulsion, steric unevenness, acceptor-acceptor collision.

Molecular docking and refinements. (A) Energies values obtained after refinement by FireDock. (B) Molecular Docking and Refinements. (A) Energy values obtained after refinement by FireDock. (B) Best 3D docking model acquired after docking in PatchDock and subsequent refinements in FireDock, HADDOCK, MD, and QM/MM (structure PFHMQM). At the bottom of the image, energetic scores of the structures following sequential stages of geometric refinement.
Codon adaptation and in silico cloning
Codon adaptation according to the host microorganism to be used is a very important step for in silico cloning. For this purpose, we used the Java Codon Adaptation Tool (http://www.jcat.de/), which specializes in predicting an optimized coding sequence for each input sequence (DNA or protein). Its result output includes the optimized gene sequence along with its codon adaptive index (CAI) and the percentage of CG content61. In this step, E. coli k12 was considered, which is widely used as a host microorganism. In addition, three criteria were selected (a) avoidance of rho-independent transcription terminators, (b) avoidance of prokaryotic ribosome binding sites, (c) avoidance of restriction enzyme cleavage sites. This aligned sequence was inverted using the IUPAC convention (https://arep.med.harvard.edu/labgc/adnan/projects/Utilities/revcomp.html) to show complementarity with the replication cycle of its vector. The restriction sites XhoI and BamHI were added to the N-terminus and C-terminus of the optimized reverse cDNA sequence. This resulting sequence was inserted into the pET-28a( +) vector using SnapGene v4.2 software (http://www.snapgene.com)62 for subsequent in silico cloning.
Immune response simulation
The immunogenicity and immune response of the vaccine construct were assessed using the C-ImmSim service (https://kraken.iac.rm.cnr.it/C-IMMSIM/), which combines molecular biology approaches with data-driven prediction methods to provide a comprehensive profile63. The program was adjusted so that the period between injection doses is approximately one month, which equates to (84 time steps), and the simulation steps were set to one thousand, while all other stimulation parameters were left at their default values.
Results and discussion
Vaccines are the best strategy to prevent infectious diseases by generating protective immunity. Conventional vaccines are used worldwide and are considered the best method for treating various diseases. However, new vaccination tactics are required immediately to address the problems associated with live or attenuated vaccines (see our introductory section). For example, multiepitope-based vaccines produced by reverse vaccinology techniques are harmless, more stable, and easier to produce than attenuated viral vaccines. In addition, they are recommended primarily for their cost-effectiveness and higher efficacy64,65.
The importance of computational methods in the development of these vaccines is growing. Current approaches in molecular modeling, bioinformatics, and immunoinformatic have accelerated the production process and enabled screening of genomes to identify potential vaccine candidates and develop multiepitope vaccines with higher efficacy. This technology has evolved to identify viral proteome areas that are potentially capable of activating innate and adaptive immune responses to induce protective memory. Therefore, it has been used in several studies with pathogenic microorganisms such as SARS-CoV-220 and other arboviruses such as dengue virus44, Burkholderia64, Chikungunya virus24, and Mayaro virus25,26. Recent studies in animal models have shown excellent results for multiepitope vaccines, suggesting that this platform is a promising and safe method compared with attenuated vaccines66. Interestingly, the first candidate vaccine against malaria to progress to phase III clinical trials is the MosquirixTM, which comprises contiguous epitopes derived from the circumsporozoite protein of Plasmodium falciparum67.
In the present research, we proposed a multi-epitope vaccine against Yellow fever virus based on a robust methodology. The results will be presented and discussed below.
Acquisition of viral protein sequences
In our quest to explore antigenic epitopes for the development of an effective Yellow Fever Virus (YFV) vaccine, we conducted a thorough examination of validated data from the ViPR database. This investigation yielded a total of 5, 5, 3, 9, 10, 10, 6, 10, 7, 5, and 4 protein sequences for the proteins E, C, M, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, respectively, within the YFV. All these proteins were taken from a total of 196 complete genomes, included sequences from different geographical regions such as Africa (27), Asia (9), Europe (28), North America (7) and South America (125). The consensus sequences of these proteins were used to predict the B and T cell epitopes for the design of the multi-epitope vaccine.
CTL epitope selection
Combining Propred and NetCTL data, 110 epitopes with binding affinity to MHC class I were identified. However, only 10,\(({M}^{60-68}\), \(NS2{A}^{105-113}\),\(NS2{B}^{5-13}\), \(NS2{B}^{8-16}\), \(NS2{B}^{9-17}\), \(NS2{B}^{79-87}\), \(NS{3}^{123-131}\), \(NS4{A}^{106-114}\), \(NS4{B}^{199-207}\) and \(NS{3}^{144-152})\), had a binding affinity of \(\ge\) 5 alleles, obtained relevant results in antigenicity, allergenicity, immunogenicity, non-toxicity, and had a maintenance rate of \(\ge\) 90% (Tables 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 and 11). One by one, we will present and analyze the results for each YFV protein.
Some studies concluded that the envelope protein is critical for eliciting a strong humoral and cellular adaptative immune response against YFV68,69. Analysis of most antigenic regions of this protein revealed a total of 13 epitopes with possible affinity for MHC-I, but epitopes \({E}^{40-48}\), \({E}^{237-245}\), \({E}^{265-273}\), and \({E}^{334-342}\) did not have binding affinity to at least 5 HLA alleles, so they were discarded from our analysis. Of the remaining 9 epitopes, 6 had no antigenicity values above 0.5, and \({E}^{234-242}\) was classified as a probable allergen. \({E}^{239-247}\) and \({E}^{284-292}\) had negative immunogenicity values and were also discarded (Table 2).
Melo et al. (2013)70 provided six epitopes of the YFV envelope protein that elicited both CD4 + and CD8 + T cells, namely \({E}^{57-71}\), \({E}^{65-79}\), \({E}^{72-87}\), \({E}^{337-351}\), \({E}^{345-359}\), and \({E}^{361-375}\)70. Milton et al.71 showed that \({E}^{57-71}\) and \({E}^{329-343}\) (\({E}^{57-71}\), \({E}^{61-75}\) , \({E}^{129-145}\) and \({E}^{135-147)}\) generate the highest CD8 + (CD4 +) T cell responses in mice71. Recently, Hassan et al.72 found that \({E}^{471-479}\), \({E}^{363-371}\), and \({E}^{226-234}\) (\({E}^{284-292}\) and \({E}^{479-487}\)) interact only with MHC-I and MHC-II alleles with extensive population coverage72.
As we saw here, none of these peptides matched our predicted peptides. Using a more robust and sophisticated methodology, we excluded \({E}^{72-87}\)70,71\({E}^{329-343}\), and \({E}^{337-351}\)70 epitopes because they had very low antigenicity values31. In addition, we discarded \({E}^{284-292}\)72 because it had an immunogenicity score (i.e., a T-cell recognition score) of -0.19211, indicating that the peptide-MHC-I complex formed by this epitope is theoretically not immunogenic in humans32. The selection of such epitopes (and many others) for the construction of a vaccine would not confer immunogenic power to it, which could logically be confirmed only by experimental testing.
It is essential to stress that we configure and parameterize the tools we use for highly sensitive analysis. Although we found a lower number of epitopes than other authors, we guarantee high confidence in the regions of the YFV proteome selected for the composition of our vaccine prototype by avoiding false positives.
The promiscuous epitope \({M}^{57-65}\), which belonged to 11 HLA class I alleles, had an antigenicity of 0.5028 but was noted as a possible allergen in the allergenicity analysis. \({M}^{39-47}\), \({M}^{41-49}\) and \({M}^{60-68}\) had satisfactory results in all analyses. However, only \({M}^{60-68}\), which was predicted for 14 alleles, was conserved in all M protein sequences (Table 3).
As happened with protein E, the epitopes of protein C did not pass through the analyses to which they were subjected, \({C}^{3-11}\), \({}^{74-82}\) \({C}^{77-85}\), not associated with the number of alleles required. Epitopes \({C}^{73-81}\), \({C}^{80-88}\), \({C}^{83-91}\) obtained antigenicity values below 0.5, remaining only \({C}^{70-78}\), which presented as a possible allergen (Table 4).
Only 2 epitopes were identified in the NS1 protein, \(NS{1}^{34-42}\) and \(NS{1}^{176-184}\), which were associated with 11 and 12 alleles, respectively, but the antigenicity values were less than 0.5, suggesting nonantigenic sequences, so they were not considered for further testing (Table 5).
The NS2A protein had a total of 19 epitopes (Table 6). \(NS2{A}^{98-106}\), \(NS2{A}^{135-143}\), \(NS2{A}^{142-151}\) and \(NS2{A}^{144-143}\) did not have the required number of alleles. Epitopes \(NS2{A}^{44-52}\), \(NS2{A}^{123-132}\) and \(NS2{A}^{195-203}\) had significant scores of 0.8864, 0.8250, and 0.819, respectively, but were classified as allergenic molecules. \(NS2{A}^{34-42}\) had a score of 0.8618 and was not allergenic, but had an immunogenic score of -0.04624, indicating non-immunogenic sequences. Epitopes \(NS2{A}^{13-21}\), \(NS2{A}^{40-48}\) and \(NS2{A}^{105-113}\), which were predicted to bind to 15, 13, and 7 alleles, respectively, performed well in terms of antigenicity, allergenicity, immunogenicity, and toxicity, but only \(NS2{A}^{105-113}\) was conserved in 90% of the sequences (Table 6).
Of the 15 epitopes of the NS2B protein that demonstrated affinity for the MHC I molecule, epitope \(NS2{B}^{72-80}\) failed to bind to \(\ge\) 5 alleles, and subsequently \(NS2{B}^{96-104}\) failed the allergenicity test. The remaining epitopes passed the other analyses, but only \(NS2{B}^{5-13}\), \(NS2{B}^{8-16}\), \(NS2{B}^{9-17}\) and \(NS2{B}^{79-87}\) had sequences conserved in all NS2B sequences used (Table 7).
The NS3 protein was able to preserve the \(NS{3}^{123-131}\) and \(NS{3}^{144-152}\) epitopes throughout the analysis. The promiscuous epitope \(NS{3}^{67-75}\) was predicted to bind only 3 HLA. \(NS{3}^{225-233}\) and \(NS{3}^{403-411}\) had significant antigenicity values but were disregarded for further analysis because they were considered allergenic. The immunogenicity values of epitopes \(NS{3}^{374-382}\) and \(NS{3}^{430-438}\) were negative and therefore had to be discarded (Table 8).
Table 9 shows that the peptide sequence \(NS4{A}^{48-56}\) was not considered for antigenicity analysis because it has binding affinity for only 4 HLA alleles. Only \(NS4{A}^{4-12}\) and \(NS4{A}^{106-114}\) were considered antigenic, but the immunogenicity value of the first peptide was significantly low. Therefore, only the \(NS4{A}^{106-114}\) epitope of NS4A protein was further analyzed and allowed in all phases.
\(NS4{B}^{21-29}\) binds to 1 HLA class I allele, and the others are linked to \(\ge\) 5 alleles. \(NS4{B}^{167-175}\) was determined to be antigenic and not-allergic, but had negative immunogenicity. Of the other 6 epitopes of NS4B protein that were classified as antigenic, non-allergic, only \(NS4{B}^{199-207}\) was found to be immunogenic, non-toxic and 100% conserved. (Table 10).
Originally, many epitopes were predicted for the NS5 protein, but none of the epitopes that were able to bind to the required number of alleles passed all tests (Table 11).
The identification of epitopes that can be recognized by TCD8 + lymphocytes is very important in the cellular immune responses against intracellular microorganisms, such as viruses46. So, we hope that these sequences can activate this response and the infected cells can be destroyed.
HTL epitope selection
Bringing together NetMHCII and NetMHCIIpan data, a total of 365 potential epitopes with possible binding affinity to the MHC class 2 were found. Nonetheless, only 9 (\(NS{3}^{47-55}\), \(NS{3}^{49-57}\), \(NS{3}^{217-225}\), \(NS{3}^{218-226}\), \(NS{3}^{267-275}\), \(NS{3}^{268-276}\), \(NS{3}^{448-456}\), \(NS{3}^{449-457}\), \(NS{5}^{398-406}\)) had binding affinity of \(\ge\) 5 alleles, showed considerable antigenicity, allergenicity, immunogenicity, non-toxicity results and possessed a conservation \(\ge\) 90% (Table 12).
BL epitope selection
The adaptive humoral immune response is one of the most sought after effects when it comes to vaccines. T-helper lymphocytes are crucial in the differentiation of B-lymphocytes. For this reason, finding LB epitopes that overlap with HTL epitopes ensures protection for different cell types working together. Here, a total of 98 BL epitopes, of varying size, were identified initially using the IEDB server. Of these, only 11 had a conservation of \(\ge\) 90% and were used for sequence overlap analysis with HTL epitopes, in which only 5 (\(NS{3}^{277-281}\), \(NS{3}^{443-458}\), \(NS{3}^{458-471}\) and \(NS{5}^{401-409}\)) found matching sequences (Table 13).
Recently, Tosta et al.73 found 28 CTL (06 HTL) epitopes overlapping with B-cell epitopes, including three (one) from the envelope, two (one) from the capsid, two from prM, four (one) from NS1, two (one) from NS2A, five from NS3, one (two) from NS4B, nine from NS573. As mentioned earlier, no epitope matches our results, as we are extremely careful to avoid false positives as much as possible.
In our study, significant progress was made in immunoinformatic analysis, highlighting crucial aspects in epitope selection for the development of vaccines against flaviviruses such as YFV and Zika virus. Through comprehensive immunoinformatic analyzes, we were able to identify important epitopes in the NS3 and NS5 proteins of these viruses. The identified epitopes “PTRVV” (277-PTRVV) and “YMWLGARYL” (477-YMWLGARYL) can be compared to the promiscuous T-cell and B-cell epitopes predicted for Zika virus by Dar et al.74. The identification of overlapping or similar epitopes in different flaviviruses such as Zika and YFV emphasizes the potential for cross-reactivity or shared immunological features between these viruses. However, “YMWLGARYL” shows intriguing complexity in epitope selection as it fails in allergenicity assays. This finding underscores the need to consider a range of factors, including allergenicity, when developing vaccines.
Population coverage
As we have seen so far, one of the most important concerns in vaccine development is the efficacy of the vaccine considering regional populations in the case of endemic diseases. Our method aims to predict overlapping epitopes between CTL, HTL, and B cells that can be recognized by the most common HLA alleles in Africa and South American populations and thus can induce both humoral and cellular immune responses.
Population coverage analysis was performed on 10 CTL and 5 HTL epitopes that overlapped with LB epitopes, seeking to identify the most common alleles in Africa and North America. The epitopes \({M}^{60-68}\), \(NS{3}^{267-275}\), \(NS{3}^{268-276}\), \(NS{3}^{448-456}\), \(NS{3}^{449-457}\), \(NS4{A}^{106-114}\) and \(NS{5}^{398-406}\) obtained a population coverage \(>\) 50%. \(NS2{A}^{105-113}\), \(NS2{B}^{5-13}\), \(NS2{B}^{8-16}\), \(NS2{B}^{9-17}\), \(NS2{B}^{79-87}\) and \(NS4{B}^{199-207}\) had a coverage percentage between 24.57% and 42.09%. Only the epitopes \(NS{3}^{123-131}\) and \(NS{3}^{144-152}\) had a coverage \(<\) 20%. Which shows that most of the final epitopes have excellent population coverage in the regions of interest (Table 14).
Vaccine sequence construction
Having identified the immunogenic, nonallergenic, nontoxic, conserved epitopes of the YFV proteome capable of binding to a substantial number of alleles of the HLA system, we propose the construction of a vaccine prototype using these epitopes. Precisely because we know that vaccines based on classical vaccine platforms can cause allergic reactions because they are formulated with multiple proteins of the target organism75, we are interested in developing a prototype with epitopes that have the most important properties of a vaccine candidate, including minimal allergic reactions, toxicity, and side effects. In addition, we used the linkers in our multi-epitope vaccine to reduce (improve) the likelihood of misfolding of the fusion epitopes and low yield in vaccine production (folding, stability, and flexibility or stiffness of the designed chimeric vaccine candidate)76. At the N-terminus of our vaccine sequence, the apeptide adjuvant \(\beta\)-defensin was added to ensure high antigenicity and enhance the immunological response38.
Thus, our prototype vaccine consists of 10 CTL and 5 HTL with overlapping LB epitopes that are bonded together with the adjuvant \(beta\)-defensin, yielding a subunit vaccine with a total of 264 amino acids (Fig. 1). In general, the development of a vaccine is quite tedious and takes a long time. However, the early stages of this work demonstrate how immunoinformatics helps to reduce the required research time by selecting only the best sequences from a substantial number of viral proteins that are able to activate humoral and cellular immune responses against YFV.
Because simply binding to the MHC complex does not guarantee that these sequences are epitopes77, only those that met the previously defined criteria were used to construct our prototype, specifically.
Physicochemical parameter prediction
The chemical formula of the vaccine sequence was predicted to be C1213H1970N348O336S10. The molecular weight was 27,125.72 kDa, the theoretical isoelectric point (pI) was 9.87, the instability index was 27.87, the aliphatic index was 100.42, and GRAVY was 0.315. These results indicate the preparation of a basic, stable (cut-off point \(<\) 40), thermostable, and hydrophobic sequence, which, with the exception of hydrophobicity, are favorable parameters for a vaccine.
Design, refinement and validation of tertiary structure of the vaccine prototype
The amino acid sequence was uploaded into the Raptor X server, and the tertiary structure of the vaccine that is based on several epitopes was constructed as a consequence. Of the 264 residues, 264 (100%) were modeled, while 61 (23%) predicted positions were disordered. Absolute model quality is measured by total uGDT (GDT), which was predicted to be 94. For a protein with \(>\) 100 residues, uGD \(>\) 50 is a good indicator78. The relative quality of the model was also evaluated and a p-value of 1.559 × 10−3 was obtained. All these results indicate a good overall tertiary model of the subunit vaccine, as the maximum number of residuals was modeled.
Vaccine model refinement was performed using the GalaxyRefine2 server. The GalaxyRefine creates 5 refined models, and model 1 was selected as the final vaccine model for further analysis because it produced the best results, including GDT-HA (0.9725), RMSD (0.342), MolProbity (1.761), Clash score (9.5), Poor rotamers (0.6), and Rama favored (96.2) (Fig. 1). A higher GDT-HA value indicates superior overall model quality. The MolProbity score serves as a critical protein quality metric, amalgamating Clashscore, Rotamer, and Ramachandran scores into a normalized single score. RMSD provides insight into the average atom deviation between the refined and unrefined structure, and ideally, it should be minimal. A lower MolProbity value often correlates with the promotion of TH2 cytokines, fostering B-cell and antibody responses47.
To assess the structural and stereochemical integrity of the refined models, comprehensive all-atom structure validation analyses were conducted. These analyses encompassed the use of the Ramachandran plot, Z-score, ERRAT, Verify3D, and PROCHECK tools. Ramachandran plot revealed that 95.83% of the residues were in the most favorable ranges, 5.17% were in allowed ranges. There is no percentage of disallowed ranges. The Z-score of the model was estimated to be − 4.26, which is within the range of scores normally found for native proteins of similar size (Fig. 3A). As per the ERRAT assessment, the protein model exhibited an overall quality factor of 87.20% when compared to highly refined structures. Furthermore, in Verify3D, 89.02% of the residues achieved a 3D-1D score of ≥ 2 (as depicted in Fig. 3B, C), indicating a high level of compatibility with the benchmark models. This validation process confirmed the tertiary structure model's integrity, with no critical errors detected.

Validation of the final subunit vaccine model. (A) Vaccine 3D Structure Validation by ProSA-web illustrating Z-score; (B) Quality factor and quality score by ERRAT (C) Verify3D tools, respectively.
The results of the Molecular Dynamics (MD) simulations, portraying the stability and flexibility of the vaccine, are illustrated in Fig. 4. The root mean square deviation (RMSD) plot initially showed very little variation up to 15 ns in the range of 0.4–0.9 nm, followed by a stable conformation up to 50 ns. This stability could be due to the higher number of stable bonds of the target protein. Then, the root mean square fluctuation (RMSF) value was calculated to investigate the structural flexibility of the backbone atoms of the protein. The results show that there are no large fluctuations and that the complex is flexible (RMSF \(\le\) 0.8 nm).

3D structural conformation of the multi-epitope subunit vaccine after homology modeling and refinement by SwissModel and Galaxy Refine servers.
MD simulations can be used prior to docking, as a set of “new” and broader protein conformations can be extracted from the processing of the resulting trajectory and used as targets for docking. These results suggest that the developed vaccines can strongly interact with immune receptors.
Molecular docking, refinement and comparative analysis
In the recognition of YFV by the host immune system, Toll-Like Receptor 2 (TLR-2) has been identified along with three other Toll-Like receptors (7, 8, and 9) as critical in the interactions between the 17D vaccine and human cells. According to Pulendran47, TLR-2 has the ability to induce both Th1 and Th2 cells and can indirectly facilitate either antibody production or a cytotoxic cellular response. Hence, the interaction between TLR-2 and the vaccine prototype was evaluated through protein–protein docking and was validated through a comprehensive structural validation protocol.
The binding of the refined vaccine model performed by the PatchDock server with TLR-2 resulted in 20 models ranked by a geometric complementarity score. The top 10 models were refined based on binding energy using FireDock (Fig. 2a). In the HADDOCK refinement process, a solvent shell was constructed around the top complexes, followed by a series of brief Molecular Dynamics (MD) simulations governed by the subsequent parameters: all atoms, excluding side-chain atoms at the interface, were restrained to their original positions. Subsequently, 1250 MD steps were executed at 300 K with position restraints applied to heavy atoms not participating in the Protein–Protein Interaction (PPI) (specifically, residues outside of intermolecular contacts within 5 Å). The systems were gradually cooled down through 1000 MD steps at 300, 200, and 100 K, while maintaining position restraints on the backbone atoms of the protein complex, except for those at the interface. In the final step, the optimal model was refined by applying the methodology of quantum mechanics/molecular mechanics (QM/MM).
In our research, we conducted a comparative analysis between structures derived from basic geometric optimization using classical mechanics, resulting in the 'PatchDock + FireDock + HADDOCK' (PFH) structure, and those obtained from advanced computational methods such as molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) techniques, which led to the ‘PatchDock + FireDock + HADDOCK + MD + QM/MM’ (PFHMQM) structure. We utilized the PRODIGY tool to assess energy scores and binding affinity of both models, as shown in Fig. 2b. This comparison unveiled significant discrepancies, particularly in the RMSD and Gibbs free energy parameters. The RMSD for PFH (6.3 Å) was higher than that for PFHMQM (2.2 Å), and ΔG_PFHMQM (− 17.1 kcal/mol) was lower compared to ΔG_PFH (− 12.9 kcal/mol). These results emphasize the importance of sophisticated geometric optimization calculations that provide a more accurate and reliable depiction of the system's electronic cloud. Consequently, even subtle structural changes brought about by these calculations can significantly alter ligand binding to receptors.
Finally, Discovery Studio, LigPlot + and PoseView were used to extract the graphical image of the vaccine-receptor interaction profile of the QM /MM complex. Of the total 28 intermolecular interactions, 14 were of hydrogen bonding type (GLU69-SER40, ALA161-SER56, ALA264-LYS252, VAL183-LYS505, GLY185-TRP529, ALA71-SER40, TYR157-ASP31, SER160-ASP58, ARG182-ASN466 , SER160-SER56, ARG182-ASN466, PRO186-GLN557, ARG195-ASP463, ARG224-GLU481), 8 strongly electrostatic in nature (ARG195-ASP463, ARG224-GLU481, ARG224-GLU481, GLU69-LYS37, ARG195-ARGSP2419-GLU481, ARG195-TYR483) and 7 with a polar/hydrophobic properties (PRO165-LYS55, ALA162-ALA80, ALA161-ILE35, LA264-LEU280, ALA264-LYS308, ALA162-HIS104, TYR157-ILE35) (Fig. 5).

Intermolecular interactions between vaccine-TLR-2 evidenced by the contact surface. (A) Hydrogen bonds. (B) Electrostatic bonds. (C) Hydrophobic bonds.
In summary, the docking analysis revealed robust interactions between the vaccine molecules and immune cells. However, it's important to note that this assessment was theoretical, and a real evaluation of binding potency within the host is still needed. To validate the docking results, various techniques, including molecular dynamics and QM/MM simulations, were employed. The MD + QM/MM analysis predicted substantial binding stability, which is vital for ensuring the lasting effectiveness and durability of the vaccine construct within the host.
Codon adaptation and in silico cloning
Validation of the candidate vaccine construct necessitates immunoreactivity screening via serological analysis32. This involves the expression of the recombinant protein in Escherichia coli expression systems, as these systems are well-suited for the production of recombinant proteins79,80. Codon optimization performed to achieve high-level expression of our vaccine prototype (801 nucleotides) in E. coli (strain K12) shows that the codon adaptability index (1.0) and GC content (55.42%) were favorable for a high-level expression in bacteria.
For gene expression in an organism of interest, the ideal CAI value should be 1.00, but a value \(>\) 0.8 is also considered good and the percent GC content should be in the range of 30–70%. These results suggest a good expression of the genetically engineered vaccine in the E. coli K-12 strain. After evaluating these parameters and once the sequence was free of commercially available restriction sites, two restriction sites XhoI and BamHI were added to the N- and C-terminal, respectively, of the optimized reverse codon sequence. Ultimately, the recombinant plasmid was generated by incorporating the reverse sequence of the adapted codon into the pET-28a( +) vector. The chosen restriction sites for this insertion were PspXI and BamHI, which served as the starting and ending cut points, respectively (Fig. 6).

In silico cloning of the final vaccine construct into the pET28a ( +) expression vector.
Immune simulation of the multiepitope vaccine
Although available immunoinformatic algorithms for in silico predictors of epitopes and potential vaccines demonstrate remarkable precision, one of the greatest hurdles in the field is to correctly stimulate the immune system81. Here, the immune response simulation results obtained using C-ImmSim revealed that the secondary immune response, overall, was significantly greater than the first response, coinciding with what occurs in vivo immune response (Fig. 7).

Graphs of the immune response elicited at the first and second dose of vaccine. (A) Antigens (Ag) and antibodies (Ac) Antibodies are divided by isotypes: IgM + IgG, IgM, IgG1 + IgG2, IgG1 and IgG2; (B) B lymphocyte (LB) population: total, memory cells, no memory (B non Mem) and IgM, IgG1 and IgG2 isotypes; (C) B lymphocyte (LB) population by state: active, internalized Ag (Internalized), presenting to MHC class II (Presenting-2), doubling and anergic; (D) T-helper lymphocyte (THL) population: total, memory cells (TH Mem) and no memory (TH non Mem); (E) T-helper lymphocyte (LTH) population by state: active, doubling, resting, and anergic; (F) T-helper lymphocyte profile; (G) T-cytotoxic lymphocyte (CTL) population: total, memory cells (TC Mem) and no memory (TC non-Mem), (H) T-cytotoxic lymphocyte population by state: active, doubling, resting and anergic; (I) Cytokine and interleukin concentration. D in the inserted graph is a danger signal; (J) Dendritic cells: internalized Ag, presenting to MHC class I and II (Presenting-1 and Presenting-2), total, active and resting, (k) Macrophages: internalized Ag, presenting to MHC class II, total, active and resting.
One of the key elements in the immune response to the Yellow Fever Virus (YFV) is the presence of neutralizing antibodies, primarily of the IgG type. These antibodies play a critical role in establishing a long-lasting immune defense against YFV and are considered a significant gauge of immunity70. Following the administration of a peptide vaccine booster, there was a notable increase in the antibody response, accompanied by a simultaneous decrease in antigen levels. This was evident from the narrower base of the antigenic spike in comparison to the previous dosage. During this period, a predominant humoral response was observed, characterized by higher IgM production. Despite the increased IgM production, the booster resulted in higher quantities of IgG compared to the initial dose, indicating a degree of seroconversion (see Fig. 7A).
Analysis of B-cell population per cell showed overall B-cell memory responses higher after boost dose and which remains in greater proportion for more than 200 days (Fig. 7B). In Fig. 7C, it is evident that the B-cell population remained higher and stable over the same time periods. As for the T cells, after the prime dose, there was a simultaneous increase in effector T-helper (Th) cells, while Th-memory cells showed a lower response, but after the boost, Th-memory respond faster and higher as expected for an efficient immune response to antigenic challenge (Fig. 7D). At the same period, there was a predominance of Th active cells (Fig. 7E). There was an increase of active effect cytotoxic T-cells after prime dose and following around day 30 (Fig. 7G, H), which corresponds to the period of application of the second dose, explaining that there was a previous recognition of these antigens.
After the initial prime dose, there was an initial surge in the IFN-γ response, which is associated with both CD8 + T-cell and CD4 + Th1 response. Additionally, there was a notable response in terms of IL-10 and TGF-β cytokines, which are associated with the T-regulatory (T-reg) phenotype. After boost, IFN-\(\gamma\) also peaked and TGF-b had a higher production than prime dose (Fig. 7I). The cytokines IL-12 and IL-2 increased in the second phase of vaccination, this increase reinforces the modulation to a Th1 cell-mediated response82, moreover, IL-12 production correlates with Natural Killer (NK) cells. NK cells are innate lymphocytes and their role is well established in the immune response against viruses83. Its function is regulated, in part, by the action of cytokines such as IL-12, which act to activate these cells. The importance of NK cells in yellow fever disease has been suggested84.
There was observed increase in dendritic cells activities throughout the duration of the simulation (Fig. 7J). Active macrophages increase after the administration of vaccine doses, being probably responsible for the antigenic presentation of the vaccine peptide (Fig. 7K). This is an important event indicating that the vaccine construct showed the ability to stimulate the right immunological compartment to effective response. Moreover, the increase in the Th1 response observed after the first and second doses are important because they demonstrate that the construct vaccine is effective in mounting a specific response to eliminate the YF virus, considering that CD4 T cells are important to activate CD8 T cells, and also for the activation of B cells, favoring affinity maturation and immunoglobulin isotype switching (Fig. 7F).
Our results confirm the findings of de Melo et al.70, who were the first to demonstrate that TCD4 + lymphocyte-recognized epitopes play a crucial role in the immune response against YFV. Helping humoral immune response (B-lymphocyte differentiation) activated TCD4 + cells may differentiate into Th1, Th2 and Th17, based on their cytokine secretion profile. This type of mixed response has already been verified with the yellow fever vaccine 17DD, including IL-2, IFN-\(\gamma\), TNF-\(\alpha\), IL-12 (TH1), IL-4, IL-5, IL-10, and IL-13 (TH2). TH2 cytokines promote B-cell and antibody responses whereas TH1 T cells habitually promote CD8 + responses47.
The multi-epitope vaccine under consideration was intended to serve as an effective vaccine and could also function as a booster in case of mutations in the YFV. An evaluation of memory T-cell responses and neutralizing antibody levels in individuals who received primary vaccines ranging from 45 to 13 years after vaccination revealed a decline in memory responses after 10 years of YFV vaccination. This decline was observed in classical memory B-cells, CD4 + and CD8 + T-cells85. To enhance memory responses, booster shots may be necessary, which could include the multi-epitope vaccine as an adjuvant peptide to provide a targeted and directed immune response against YFV.
Conclusion
In summary, we identified 15 epitopes for T and B cells in the structural and nonstructural proteins of 196 strains of YFV that have essential properties for eliciting an effective and population-wide immune response, namely high antigenicity and immunogenicity, extensive conservation among different strains, comprehensive population coverage, non-toxicity and non-allergenic in humans. On this basis, we have developed and optimized the spatial geometry of a prototype subunit vaccine with appropriate physicochemical properties, the ability to bind to the Toll-2 receptor, the potential to elicit an effective and durable immune response in two doses, and a coding region that can be successfully inserted into a cloning vector. Experimental studies (in vitro and in vivo) are required to validate the efficacy and safety of the proposed vaccine (Supplementary information 1).
Data availability
All data generated or analysed during this study are included in this published article.
References
Mutebi, J.-P., Wang, H., Li, L., Bryant, J. E. & Barrett, A. D. Phylogenetic and evolutionary relationships among yellow fever virus isolates in Africa. J. Virol. 75(15), 6999–7008 (2001).
Wang, E. et al. Genetic variation in yellow fever virus: duplication in the 3’ noncoding region of strains from Africa. Virology 225(2), 274–281 (1996).
Strode, G. K. Yellow Fever (McGraw-Hill, 1951).
Figueiredo, L. T. M. Febres hemorrágicas por vírus no Brasil. Revista da Sociedade Brasileira de Medicina Tropical 39, 203–210 (2006).
da Vasconcelos, P. F. C. Febre amarela: reflexões sobre a doença, as perspectivas para o século XXI e o risco da reurbanização. Revista Brasileira de Epidemiologia 5, 244–258 (2002).
Vellozzi, C. et al. Yellow fever vaccine-associated viscerotropic disease (YEL-AVD) and corticosteroid therapy: Eleven United States cases, 1996–2004. Am. J. Trop. Med. Hyg. 75(2), 333–336 (2006).
De Nishioka, A. S., Nunes-Ara£jo, F., Pires, W. P., Silva, F. A. & Costa, H. L. Yellow fever vaccination during pregnancy and spontaneous abortion: a case-control study. Trop. Med. Int. Health 3(1), 29–33 (1998).
Gotuzzo, E., Yactayo, S. & Córdova, E. Efficacy and duration of immunity after yellow fever vaccination: Systematic review on the need for a booster every 10 years. Am. J. Trop. Med. Hyg. 89(3), 434 (2013).
Khromava, A. Y. et al. Yellow fever vaccine: An updated assessment of advanced age as a risk factor for serious adverse events. Vaccine 23(25), 3256–3263 (2005).
Lindsey, N. P. et al. Adverse event reports following yellow fever vaccination. Vaccine 26(48), 6077–6082 (2008).
Organization WH, others. Vaccines and vaccination against yellow fever: WHO position paper—June 2013. Weekly Epidemiological Record= Relevé épidémiologique hebdomadaire 2013;88(27):269–283.
Rafferty, E., Duclos, P., Yactayo, S. & Schuster, M. Risk of yellow fever vaccine-associated viscerotropic disease among the elderly: A systematic review. Vaccine 31(49), 5798–5805 (2013).
Domingo, C., Charrel, R. N., Schmidt-Chanasit, J., Zeller, H. & Reusken, C. Yellow fever in the diagnostics laboratory. Emerg. Microbes Infect. 7(1), 1–15 (2018).
da Vasconcelos, P. F. C. et al. Serious adverse events associated with yellow fever 17DD vaccine in Brazil: A report of two cases. Lancet 358, 91–97 (2001).
Lawrence, G. L., Burgess, M. A. & Kass, R. B. Age-related risk of adverse events following yellow fever vaccination in Australia. Commun. Dis. Intell. Q. Rep. 28(2), 244–248 (2004).
Biscayart, C. et al. Yellow fever vaccine-associated adverse events following extensive immunization in Argentina. Vaccine 32(11), 1266–1272 (2014).
Chan, R. C. et al. Hepatitis and death following vaccination with 17D–204 yellow fever vaccine. Lancet 358(9276), 121–122 (2001).
Reinhardt, B., Jaspert, R., Niedrig, M., Kostner, C. & L’age-Stehr, J. Development of viremia and humoral and cellular parameters of immune activation after vaccination with yellow fever virus strain 17D: A model of human flavivirus infection. J. Med. Virol.gy 56(2), 159–167 (1998).
Jennings, A. et al. Analysis of a yellow fever virus isolated from a fatal case of vaccine-associated human encephalitis. J. Infect. Dis. 169(3), 512–518 (1994).
de Oliveira Campos, D. M. et al. Exploiting reverse vaccinology approach for the design of a multiepitope subunit vaccine against the major SARS-CoV-2 variants. Comput. Biol. Chem. 101, 107754 (2022).
Dorosti, H. et al. Vaccinomics approach for developing multi-epitope peptide pneumococcal vaccine. J. Biomol. Struct. Dyn. 37, 3524–3535 (2019).
Ul-Rahman, A. & Shabbir, M. A. B. In silico analysis for development of epitopes-based peptide vaccine against Alkhurma hemorrhagic fever virus. J. Biomol. Struct. Dyn. 38(10), 3110–3122 (2020).
Murphy, D., Reche, P. & Flower, D. R. Selection-based design of in silico dengue epitope ensemble vaccines. Chem. Biol. Drug Des. 93(1), 21–28 (2019).
Anwar, S., Mourosi, J. T., Khan, M. F. & Hosen, M. J. Prediction of epitope-based peptide vaccine against the chikungunya virus by immuno-informatics approach. Curr. Pharmaceut. Biotechnol. 21(4), 325–340 (2020).
Silva, M. K. et al. Identification of promiscuous T cell epitopes on Mayaro virus structural proteins using immunoinformatics, molecular modeling, and QM: MM approaches. Infect. Genetics Evol. 91, 104826 (2021).
da Silva, M. K. et al. Computational vaccinology guided design of multi-epitope subunit vaccine against a neglected arbovirus of the Americas. J. Biomol. Struct. Dyn. 41, 3321–3338 (2022).
de Campos, O. D. M., Fulco, U. L., de Oliveira, C. B. S. & Oliveira, J. I. N. SARS-CoV-2 virus infection: Targets and antiviral pharmacological strategies. J. Evid. Based Med. 13, 255–260 (2020).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9), 1189–1191 (2009).
Wang, P. et al. Peptide binding predictions for HLA DR DP and DQ molecules. BMC Bioinform. 11(1), 1–12 (2010).
Jespersen, M. C., Peters, B., Nielsen, M. & Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucl. Acids Res. 45(W1), W24–W29 (2017).
Doytchinova, I. A. & Flower, D. R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8(1), 1–7 (2007).
Calis, J. J. et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 9(10), e1003266 (2013).
Vita, R. et al. The immune epitope database (IEDB) 30. Nucl. Acids Res. 43(D1), D405–D412 (2015).
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., Consortium OSDD, et al. In silico approach for predicting toxicity of peptides and proteins. PloS one 8(9), e73957 (2013).
Bui, H.-H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7(1), 1–5 (2006).
Hajighahramani, N. et al. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genetics Evol. 48, 83–94 (2017).
Kadam, A., Santanu, S. & Saudagar, P. Computational design of a potential multi-epitope subunit vaccine using immunoinformatics to fight Ebola virus. Infect. Genetics Evol. 85, 104464 (2020).
Mohan, T., Sharma, C., Bhat, A. A. & Rao, D. Modulation of HIV peptide antigen specific cellular immune response by synthetic α-and β-defensin peptides. Vaccine 31(13), 1707–1716 (2013).
Arai, R., Ueda, H., Kitayama, A., Kamiya, N. & Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 14(8), 529–532 (2001).
Gasteiger, E. et al. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook 571–607 (Humana Press, 2005).
Wiederstein, M. & Sippl, M. J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl. Acids Res. 35(suppl_2), W407–W410 (2007).
Colovos, C. & Yeates, T. O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 2(9), 1511–1519 (1993).
Eisenberg, D., Lüthy, R. & Bowie, J. U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 (1997).
Ali, M. et al. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 7(1), 1–13 (2017).
Pandey, R. K., Sharma, D., Bhatt, T. K., Sundar, S. & Prajapati, V. K. Developing imidazole analogues as potential inhibitor for Leishmania donovani trypanothione reductase: Virtual screening, molecular docking, dynamics and ADMET approach. J. Biomol. Struct. Dyn 33(12), 2541–2553 (2015).
Abbas, A. K., Lichtman, A. H. & Pillai, S. Cellular and Molecular Immunology (Elsevier, 2014).
Pulendran, B. Learning immunology from the yellow fever vaccine: innate immunity to systems vaccinology. Nat. Rev. Immunol. 9(10), 741–747 (2009).
Duhovny, D., Nussinov, R., Wolfson, H.J. Efficient unbound docking of rigid molecules. In: International workshop on algorithms in bioinformatics. Springer, 185–200 (2002).
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucl. Acids Res. 33(suppl_2), W363–W367 (2005).
Andrusier, N., Nussinov, R. & Wolfson, H. J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 69(1), 139–159 (2007).
Van Zundert, G. et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428(4), 720–725 (2016).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Andersson, M. P., Jones, M. N., Mikkelsen, K. V., You, F. & Mansouri, S. S. Quantum computing for chemical and biomolecular product design. Curr. Opin. Chem. Eng. 36, 100754 (2022).
Kar, R. K. Benefits of hybrid QM/MM over traditional classical mechanics in pharmaceutical systems. Drug Discov. Today 28, 103374 (2022).
de Sousa, B. G. et al. Molecular modelling and quantum biochemistry computations of a naturally occurring bioremediation enzyme: Alkane hydroxylase from Pseudomonas putida P1. J. Mol. Graph. Modell. 77, 232–239 (2017).
Conductance of single microRNAs chains related to the autism spectrum disorder - IOPscience. https://iopscience.iop.org/article/https://doi.org/10.1209/0295-5075/107/68006 (accessed 18 Jan 2024).
Bezerril, L. M. et al. Charge transport in fibrous/not fibrous α3-helical and (5Q, 7Q) α3 variant peptides. Appl. Phys. Lett. 98(5), 053702 (2011).
De Medeiros, A. S. A. et al. Supramolecular aggregates of oligosaccharides with co-solvents in ternary systems for the solubilizing approach of triamcinolone. Carbohydr. Polym. 151, 1040–1051 (2016).
Vangone, A. & Bonvin, A. M. Contacts-based prediction of binding affinity in protein–protein complexes. elife 4, e07454 (2015).
Mariano, D. et al. BIOINFO–Revista Brasileira de Bioinformática e Biologia Computacional (Alfahelix Publicações, 2021).
Pandey, R. K., Bhatt, T. K. & Prajapati, V. K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 8(1), 1–11 (2018).
Goldberg, M. F. et al. Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity 49(6), 1090–1102 (2018).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PloS One 5(4), e9862 (2010).
Shahab, M., Hayat, C., Sikandar, R., Zheng, G. & Akter, S. In silico designing of a multi-epitope vaccine against Burkholderia pseudomallei: Reverse vaccinology and immunoinformatics. J. Genetic Eng. Biotechnol. 20(1), 1–12 (2022).
Akter, S. et al. Immunoinformatics approach to epitope-based vaccine design against the SARS-CoV-2 in Bangladeshi patients. J. Genetic Eng. Biotechnol. 20(1), 1–14 (2022).
Vakili, B. et al. A new multi-epitope peptide vaccine induces immune responses and protection against Leishmania infantum in BALB/c mice. Med. Microbiol. Immunol. 209(1), 69–79 (2020).
Clinical, R. S. & Partnership, T. Efficacy and safety of RTS, S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 386(9988), 31–45 (2015).
Maciel, M. Jr. et al. Comprehensive analysis of T cell epitope discovery strategies using 17DD yellow fever virus structural proteins and BALB/c (H2d) mice model. Virology 378(1), 105–117 (2008).
Tottey, S. et al. Plant-produced subunit vaccine candidates against yellow fever induce virus neutralizing antibodies and confer protection against viral challenge in animal models. Am. J. Trop. Med. Hyg. 98(2), 420 (2018).
de Melo, A. B. et al. T-cell memory responses elicited by yellow fever vaccine are targeted to overlapping epitopes containing multiple HLA-I and-II binding motifs. PLoS Neglect. Trop. Dis. 7(1), e1938 (2013).
Maciel, M. Jr. et al. A DNA vaccine against yellow fever virus: development and evaluation. PLoS Neglect. Trop. Dis. 9(4), e0003693 (2015).
Hassan, H. A., Abdelrahman, K. A., Nasr, N. M. & Almofti, Y. A. Identification of novel vaccine candidates against yellow fever virus from the envelope protein: An insilico approach. J. Microbiol. Infect. Dis. 10(01), 31–46 (2020).
de Tosta, S. F. O. et al. Multi-epitope based vaccine against yellow fever virus applying immunoinformatics approaches. J. Biomol. Struct. Dyn. 39(1), 219–235 (2021).
Dar, H. et al. Prediction of promiscuous T-cell epitopes in the Zika virus polyprotein: An in silico approach. Asian Pac. J. Trop. Med. 9(9), 844–850 (2016).
McNeil, M. M. & DeStefano, F. Vaccine-associated hypersensitivity. J. Allergy Clin. Immunol. 141(2), 463–472 (2018).
Chen, X., Zaro, J. & Shen, W.-C. Fusion protein linkers: effects on production, bioactivity, and pharmacokinetics. In Fusion Protein Technologies for Biopharmaceuticals: Applications and Challenges 57–73 (Willey, 2013).
Patronov, A. & Doytchinova, I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 3(1), 120139 (2013).
Patel, S. M., Koringa, P. G., Reddy, B. B., Nathani, N. M. & Joshi, C. G. In silico analysis of consequences of non-synonymous SNPs of Slc11a2 gene in Indian bovines. Genomics Data 5, 72–79 (2015).
Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 30(5), 1102–1107 (2012).
Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 10(5), 411–421 (1999).
Six, A., Bellier, B., Thomas-Vaslin, V. & Klatzmann, D. Systems biology in vaccine design. Microb. Biotechnol. 5(2), 295–304 (2012).
Spellberg, B. & Edwards, J. E. Jr. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 32(1), 76–102 (2001).
Lodoen, M. B. & Lanier, L. L. Natural killer cells as an initial defense against pathogens. Curr. Opin. Immunol. 18(4), 391–398 (2006).
Marquardt, N. et al. The human NK cell response to yellow fever virus 17D is primarily governed by NK cell differentiation independently of NK cell education. J. Immunol. 195(7), 3262–3272 (2015).
Azevedo, A.C.C., Pereira, C.C., do Antonelli, L.R.V., Fonseca, C.T., Carvalho, A.T., Rezende, G.V., et al. Booster dose after 10 years is recommended following 17DD-YF primary vaccination. (2016).
Acknowledgements
The authors, Thanks to the Brazilian Research Agencies CAPES and CNPq. The authors extend their appreciation to the Researchers Supporting Project number (RSP2024R197), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, OLTDS., MKDS., HAN and JFRN., methodology, MB, OLTDS., MKDS., and S.A. formal analysis, S.A., JPMLS and OLTDS., writing—original draft preparation, OLTDS., MKDS., SA, and JFRN. writing—review and editing, ULF, and JINO; visualization, S.A. and JFRN., supervision, JINO. funding acquisition, TMD, BS, and SA. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, O.L.T., da Silva, M.K., Rodrigues-Neto, J.F. et al. Advancing molecular modeling and reverse vaccinology in broad-spectrum yellow fever virus vaccine development. Sci Rep 14, 10842 (2024). https://doi.org/10.1038/s41598-024-60680-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-60680-9
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
