
Abstract
Markers that predict response and resistance to chimeric antigen receptor (CAR) T cells in relapsed/refractory multiple myeloma are currently missing. We subjected mononuclear cells isolated from peripheral blood and bone marrow before and after the application of approved B cell maturation antigen-directed CAR T cells to single-cell multiomic analyses to identify markers associated with resistance and early relapse. Differences between responders and nonresponders were identified at the time of leukapheresis. Nonresponders showed an immunosuppressive microenvironment characterized by increased numbers of monocytes expressing the immune checkpoint molecule CD39 and suppressed CD8+ T cell and natural killer cell function. Analysis of CAR T cells showed cytotoxic and exhausted phenotypes in hyperexpanded clones compared to low/intermediate expanded clones. We identified potential immunotherapy targets on CAR T cells, like PD1, to improve their functionality and durability. Our work provides evidence that an immunosuppressive microenvironment causes resistance to CAR T cell therapies in multiple myeloma.
Similar content being viewed by others
Main
Chimeric antigen receptor (CAR) T cells revolutionized the treatment of relapsed and refractory multiple myeloma (RRMM). Although the introduction of idecabtagene vicleucel (Ide-cel) and ciltacabtagene autoleucel (Cilta-cel) has led to unprecedented rates of deep, long-lasting remissions in heavily pretreated individuals compared to conventional therapies1, approximately 20% of individuals do not respond to Ide-cel, and the majority of individuals with suboptimal responses relapse within the first year2. Although rates of sustained measurable residual disease negativity are higher with Cilta-cel, approximately 40% of individuals show progressive disease within the first 2 years (refs. 3,4). Modes of resistance or factors predicting response to B cell maturation antigen (BCMA)-directed CAR T cells are currently missing. Biallelic loss of BCMA on chromosome 16 has been described as a tumor escape mechanism in a few case reports but does not explain relapse in the majority of individuals5,6. Due to worldwide limited availability, the considerable financial burden and the increasing number of eligible individuals for cellular therapies7, it is necessary to characterize modes of resistance and develop markers that predict responses to CAR T cell therapy. Exhaustion of CAR T cells and non-CAR host immunity, development of antidrug antibodies and shedding of BCMA through γ-secretases are further possible modes of resistance8. The dismal outcomes of individuals with extramedullary disease (EMD) and the increased incidence of EMD after relapse from CAR T cell therapy suggest that a nonpermissive microenvironment could be another important factor associated with resistance9.
Single-cell RNA sequencing (scRNA-seq) has been successfully used to study cellular dynamics associated with response to CD19-directed CAR T cell therapy in large B cell lymphoma and acute lymphoblastic leukemia10,11,12. Furthermore, scRNA-seq has shed new light on the evolution of asymptomatic plasma cell disorders over symptomatic disease to RRMM13,14,15,16,17. scRNA-seq offers the unique opportunity to study the interplay between CAR T cells, malignant plasma cells and nonmalignant cells of the host immune microenvironment. Therefore, we performed a longitudinal single-cell multiomics study to identify factors associated with response to BCMA-directed CAR T cells. By combining scRNA-seq with single-cell T cell antigen receptor (TCR) and B cell receptor (BCR) sequencing as well as single-cell surface proteomics, we aimed at identifying markers that predict response and resistance to CAR T cells in RRMM.
We found quantitative and qualitative differences in the cellular composition between responders and nonresponders at the time of leukapheresis and demonstrate that response is associated not only with the expansion of CAR T cells but also with preinfusional changes in the host immune microenvironment.
Results
Single-cell dissection of response to anti-BCMA CAR T cells
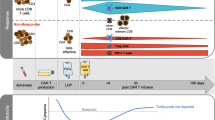
We performed single-cell RNA, BCR and TCR sequencing and single-cell surface proteomics using mononuclear cells collected at multiple time points from ten heavily pretreated individuals (median prior lines: n = 4, range 2–10; Supplementary Table 1) with RRMM treated with approved BCMA-directed CAR T cells (Ide-cel, n = 8; Cilta-cel, n = 2). Response assessment performed at day 30 after CAR T cell infusion showed complete response (CR) in five individuals and suboptimal response (very good partial remission or worse) in five individuals. Participants in the latter group showed progressive disease within the first 4 months after CAR T cell therapy (Fig. 1a). All participants were triple-class exposed, that is, they had received an immunomodulatory drug, proteasome inhibitor and anti-CD38 antibody (Fig. 1b) and had received multiple lines of prior therapies (Fig. 1c). Cytokine release syndrome was predominantly not severe (grade 1 or 2), occurred 7–11 days after infusion and required application of tocilizumab in two cases.

a, Progression-free survival in the entire cohort was significantly different when comparing individuals showing a CR with those achieving suboptimal responses using log-rank statistics. b, Summary of treatment exposure. All participants were triple-class exposed and had received an autologous stem cell transplantation. Triple-class exposure was defined by prior treatment with a proteasome inhibitor (PI; bortezomib, carfilzomib or ixazomib), an immunomodulatory drug (IMiD; thalidomide, lenalidomide or pomalidomide) and anti-CD38 antibody (daratumumab or isatuximab). Only participant P12 had received prior anti-BCMA therapies (teclistamab and belantamab mafodotin), and two participants (P11 and P14) had a history of allogenic (Allo) stem cell transplantation (SCT). CR was detected in five individuals (P7, P8, P9, P12 and P14); Auto, autologous. c, A median of four prior lines of therapy was applied; AutoSCT, autologous stem cell transplantation; AlloSCT, allogenic stem cell transplantation.
To assess longitudinal cellular dynamics of (CAR) T cells, peripheral blood mononuclear cells (PBMCs) were collected on the day of leukapheresis, on the day of reinfusion after lymphodepletion (day 0) and on days 7, 14, 30 and 100 following CAR T cell therapy (Fig. 2a summarizes sample collection and workflow). Bone marrow aspirates to isolate bone marrow mononuclear cells (BMMCs) were performed on day 30 after reinfusion. Samples collected longitudinally on the day of leukapheresis and on day 30 following CAR T cell infusion (n = 25) were subjected to single-cell multiomic analyses. In total, 179,876 cells were sequenced (median of 8,018 cells per sample, range of 1,559–11,085 cells), and 97% of cells passed quality assessment (Supplementary Table 2 summarizes the quality control data). For further analyses, participants were grouped based on their outcome into two different groups (CR versus non-CR 30 days after CAR T cell therapy).

a, Summary of workflow and sample availability. Peripheral blood (PB) samples from individuals with RRMM were collected at apheresis and days 0, 7, 14, 30 and 100 after CAR T cell infusion. Fresh peripheral blood samples were immunophenotyped by flow cytometry to track CAR and non-CAR T cells. PBMCs were isolated from fresh peripheral blood samples using Ficoll. For in vitro assays, PBMCs were transferred to CD3-coated culture plates and co-stimulated with an antibody to CD28 to induce T cell proliferation. BMMCs isolated from aspirates performed on day 30 after CAR T cell infusion were subjected to single-cell multiomic analyses alongside PBMCs collected on the same day as apheresis. Single-cell multiomics consisted of RNA, TCR and BCR sequencing as well as surface proteomics. The latter was realized by using a custom panel of 61 oligonucleotide-bound antibodies. b, Approximately 127,000 cells from 25 samples were embedded into a two-dimensional space by uniform manifold approximation and projection (UMAP). Each dot represents a single cell. Cells are colored according to cell identity. c, Bar graphs show summary statistics of the composition of cell types for each sample and the number of cells. Cell-type color codes are consistent with those in a. d, UMAP view of cell densities in individuals with a CR and those without a CR. High relative cell density is shown as dark red. e, Feature plots showing the expression (centered log ratio normalized) of lineage-specific surface antigens (for example, CD3/CD8 in CD8+ T cells or CD11/CD14/CD33 in monocytes). Cell-type abbreviations: Mono, monocyte; cDC, classical dendritic cell; pDC, plasmacytoid dendritic cell. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Analysis of bone marrow and peripheral blood after infusion
Combining single-cell transcriptomic data and tags derived from a panel of 61 oligonucleotide-bound antibodies (antibody-derived tags (ADT); Supplementary Table 3) using the weighted nearest neighbor (WNN) method18 allowed the identification of distinct immune cells (Fig. 2b) in pre- and post-CAR samples (Fig. 2c) based on achieving a CR (Fig. 2d) and the expression of canonical lineage markers (Fig. 2e). We used speckle to assess compositional changes in the CAR– T cell host immune microenvironment19.
We first investigated gene expression in different cell types isolated from bone marrow (Fig. 3a) and peripheral blood (Fig. 3b) as well as enriched gene sets in T cells (Fig. 3c) and cell-type frequencies (Fig. 3d) in the respective compartments after CAR T cell infusion (Fig. 3 and Extended Data Fig. 1). Despite the higher numbers of bone marrow plasma cells in nonresponders, individuals that demonstrated a CR harbored more CD8+ T cells, monocytes and granulocyte–monocyte progenitors in the bone marrow. Analysis of differentially expressed genes (DEGs; false discovery rate (FDR) < 0.01; Supplementary Table 4) and enriched gene sets between individuals with a CR and those without a CR demonstrated decreased activation of T and natural killer (NK) cells as well as monocytes in individuals with suboptimal responses.

a,b, Differential gene expression analysis in postinfusional BMMCs (a) and PBMCs (b) comparing nonresponders (non-CR) with responders (CR). Only cell types with significant changes in gene expression are shown (FDR < 0.05 and fold change (FC) of <1.5 or >1.5). A positive log fold change indicates upregulation in non-CR individuals. c, Gene ontology (GO) term enrichment analysis of DEGs (non-CR versus CR) for BMMCs and PBMCs. Terms are ranked by rich factor, which is the number of DEGs in the term divided by the number of background genes in that term. The dot plot depicts the most enriched GO terms for biological processes (FDR < 0.05). The dot size indicates the z score, which is the number of DEGs with a log fold change of >0 minus the number of DEGs with a log fold change of <0 divided by the square root of the number of term-associated genes. Gray/white dots indicate the same number of genes with a log fold change of >0 and <0. d,e, Comparison of cell types in CR (BMMCs n = 4; PBMCs n = 4) and non-CR (BMMCs n = 2; PBMCs n = 5). Differences between cell-type proportions were evaluated using the speckle R package based on arcsin square root transformation. Significant differences (unadjusted P values) were estimated using empirical Bayes moderated t-statistics (two sided) implemented in the speckle package (*P = 0.1, **P = 0.05, ****P = 0.001, *****P < 0.0001). The exact P values for d are as follows: plasmablast (P = 0.00002), CD14+ Mono (P = 0.06040), CD8+ T cell (P = 0.07024) and granulocyte–monocyte progenitor (GMP; P = 0.03089). The exact P values for e are as follows: CD8+ T cell (P = 0.05277), NK CD56bright (P = 0.02657) and pDC (P = 0.00060). The center of the box plots indicates the median, and the upper and lower bounds of the boxes indicate first and third interquartile ranges. Whiskers extend to 1.5× interquartile range. The following are the numbers of biologically independent samples for the contrasts: postinfusional BMMCs, n = 4 for CR and n = 2 for non-CR; postinfusional PBMCs, n = 4 for CR and n = 5 for non-CR. Abbreviations: LMPP, lymphoid-primed multipotent progenitor; HSC, hematopoietic stem cell.
Next, we analyzed paired PBMC samples collected on the same day as bone marrow aspirates after CAR T cell infusion (median of 30 days, range of 24–79 days; Fig. 3). Participants in CR (Fig. 3e) harbored fewer CD56bright NK cells and plasmacytoid dendritic cells and more CD8+ T cells than individuals with suboptimal responses (Fig. 3e). Importantly, enriched gene sets in the T cell compartment were consistent among the two analyzed compartments (peripheral blood and bone marrow; for example, cell killing and response to interferon-γ; Fig. 3c and Extended Data Fig. 1).
To investigate whether CAR T cell therapy induced not only quantitative but also qualitative changes in cellular composition, we further compared differences in cell-type composition and gene expression before and after CAR T cell infusion in bystander cells of the host immune system (Extended Data Fig. 2 and Supplemental Table 3). In nonresponders, the number of CD14+ monocytes reduced significantly after CAR T cell infusion (Extended Data Fig. 2a), and genes associated with decreased activation were enriched in monocytes and NK cells (Extended Data Fig. 2b,c). Next, we compared post- and preinfusional PBMCs in individuals with a CR (Extended Data Fig. 2 and Supplementary Table 4). Changes before and after infusion occurred in CD8+ and CD4+ non-CAR T cells (Extended Data Figs. 2b,c and 3). Gene set enrichment analysis revealed that the respective changes were associated with activation of the T cell response.
Differences between responders and nonresponders at apheresis
After investigating postinfusional peripheral blood and bone marrow samples and identifying significant changes especially in host immunity of individuals in CR, we characterized preinfusional PBMCs collected at the time of leukapheresis (Fig. 4). Individuals in CR (Fig. 4a) harbored more CD8+ T cells and NK cells but fewer CD14+ monocytes than nonresponders. Analysis of DEGs and surface protein expression also showed distinct cellular phenotypes associated with dismal outcomes after CAR T cell infusion. Consistent higher expression of genes encoding PIM kinases was detected in CD14+ and CD16+ monocytes as well as in dendritic and NK cells isolated from nonresponders at the time of leukapheresis (Fig. 4b and Supplementary Table 4). Compared to individuals in CR after therapy, enrichment of genes connected to impaired immune function was detected in CD16+ monocytes and NK cells (Fig. 4c). Beyond altered gene expression, we detected (among others) significantly higher expression of surface immune checkpoints on monocytes (for example, CD39; Fig. 4d). Remarkably, nonresponders who showed numerically lower numbers of CD8+ T cells as well as functionally impaired monocytes and NK cells had received either lymphotoxic bendamustine or an elotuzumab-based bridging therapy. In nonresponders, cell–cell interaction analyses revealed upregulated communication between monocytes and NK cells as well as plasmablasts (Fig. 4e).

a, Differences in cell-type composition between CR (n = 5 individuals) and non-CR (n = 5 individuals) in preinfusional PBMCs. Significant differences (unadjusted P values) were estimated using empirical Bayes moderated t-statistics (two sided) implemented in the speckle R package (**P = 0.05, ****P = 0.001). The exact P values are as follows: CD14+ Mono (P = 0.00086), NK (P = 0.01962) and CD8+ T cell (P = 0.01952). The center of the box plots indicates the median, and the upper and lower bounds of the boxes indicate first and third interquartile ranges. Whiskers extend to 1.5× interquartile range. b, Differential gene expression analysis in preinfusional PBMCs comparing non-CR with CR. A positive log fold change indicates upregulation in non-CR. c, GO term enrichment analysis of DEGs (non-CR versus CR). d, Analysis of surface protein expression demonstrated significantly higher expression levels of the immunosuppressive ATP ectonucleotidase CD39 on monocytes. e, Dissection of cell–cell interactions based on DEGs from b. The plot depicts potential ligand–receptor interactions between source and sender (top) and target and receiver (bottom) cell types. Dots are colored by the log fold change (LFC) directions of the DEGs. For example, ‘L:dn|R:dn’ means that the ligand (L) and receptor (R) are significantly downregulated (dn) when comparing non-CR to CR. Abbreviation: HSPC, hematopoietic stem and progenitor cell.
In vitro cytotoxicity and in vivo expansion of CAR T cells
Because we detected significant immunological differences between responders and nonresponders at leukapheresis, we aimed at characterizing the functionality of manufactured CAR T cells (Fig. 5). Under the hypothesis that the immunosuppressive microenvironment before manufacturing might lead to functionally impaired CAR T cells, we isolated untransfected T cells as well as CAR T cells 7 days after reinfusion (Fig. 5a). After CD3/CD28 stimulation and expansion, (CAR) T cells were cocultured with BCMA-expressing MM cells (U266) for 24 h (Fig. 5b). In comparison to nontransfected T cells, CAR T cells isolated at day 7 from both responders and nonresponders were capable of eliminating target cells. However, there were no significant differences in plasma cell viability between responders and nonresponders after 24 h (Fig. 5c).

a, Nontransfected T cells and CAR T cells were isolated from the peripheral blood of responders (yellow) and nonresponders (blue) on days 0 and 7 after CAR T cell infusion, respectively. Samples were ficollized and transferred to CD3-coated culture plates. T cell expansion was stimulated using an antibody to CD28. b, In vitro toxicity of CAR T cells from nonresponders and responders was assessed by elimination of a BCMA-expressing target myeloma cell line (U266). c, After a 24-h incubation, CAR T cells from responders (n = 5 individuals) and nonresponders (n = 5 individuals) were capable of eliminating target cells compared to nontransfected T cells. Statistical significance between the two groups was estimated using a Wilcoxon rank-sum test (two sided). d, Expansion of total T cells and CD4+CD8+ (CAR) T cells on days 0, 7, 14, 30 and 100 after lymphodepletion and CAR T cell infusion was measured by flow cytometry using a BCMA-labeled antibody; APC, antigen-presenting cell. e, Following lymphodepletion (LDP), T cells expanded in responders (n = 5 participants) and nonresponders (n = 5 participants). This increase was predominantly caused by expanding CD8+ T cells and was pronounced in responders. Statistical significance between the groups was estimated using a Kruskal–Wallis test. f, Analysis of CAR T cell dynamics confirmed expansion of CD8+ CAR T cells and a significant decline at day 100 after infusion in both responders and nonresponders. Only a single individual (participant P12 treated with Ide-cel) showed hyperexpanded CAR T cell clones at day 100. For c and e, unadjusted P values are shown. The center of the box plots indicates the median, and the upper and lower bounds of the boxes indicate first and third interquartile ranges. Whiskers extends to 1.5× interquartile range.
Because there were no differences in CAR T cell functionality between both groups in vitro, we tracked (CAR) T cell dynamics for the first 100 days after reinfusion using a biotin-labeled BCMA CAR detection antibody (Fig. 5d). Lymphodepletion was followed by the expansion of T cells in both groups that was primarily driven by the expansion of CD8+ cells (Fig. 5e). This marked increase of CD8+ T cells was observed not only in the CAR T cell compartment but also in nontransfected CD8+ T cells, especially in responders (Fig. 5f).
Single-cell characterization of (CAR) T cells
The expansion and persistence of CAR T cells has been associated with beneficial outcome in RRMM2. Because we detected significant differences in CD8+ and CD4+ T cell dynamics before and after CAR T cell infusion, we characterized subsets of T cells (Fig. 6a and Supplementary Table 5) and the TCR repertoire in responders and nonresponders (Fig. 6). For this purpose, sequences of Ide-cel and Cilta-cel were added to the reference genome, and Ide-cel+Cilta-cel+ CAR T cells were extracted in silico (Fig. 6b). CAR+ T cells were predominantly CD8+ in the bone marrow and peripheral blood of individuals with hyperexpanded CAR T cells after day 100 (for example, participant P12) as well as individuals with only a few detectable CAR T cells 100 days after infusion (for example, participant P14; Fig. 6b). Analysis of TCR clonality (Fig. 6c) showed that individuals in CR harbored clonotypes with higher counts more frequently at all time points and in both analyzed compartments (Fig. 6d). Although virtually no large (20 < x ≤ 100 cells) and hyperexpanded (>100 cells) clones were observed in the CD4+ T cell compartment (Fig. 6d), hyperexpanded terminally differentiated effector memory (TEMRA), cycling and exhausted CD8+ T cells occurred especially in responders after treatment (Fig. 6e). A TCR diversity analysis revealed no significant differences between both groups in CD4+ T cells and a trend toward a more diverse TCR repertoire of CD8+ T cells at the time of leukapheresis in responders (Fig. 6f). Longitudinal tracking of TCRs revealed the parallel existence of CAR+ and CAR– T cells of the same clonotype regardless of whether CAR+ T cells expanded beyond day 100 after infusion (Fig. 6g) or not (Fig. 6h).

a,b, Roughly 31,000 T cells from 25 samples were embedded into a two-dimensional space. Cells are colored by cell identity (a) and by CAR+ cells (b). The proportions of cell types for CAR+ cells in each participant (if available) are shown in b. The numbers above the bar chart represent the numbers of CAR+ cells. c, Cells were placed into clonotype groups based on cell number. d, Scatter plots between the number of T cell clonotypes and the size of the clonotype that is the number of T cells contributing to the clonotype are shown for pre- and postinfusional PBMCs (PBMC|Pre and PBMC|Post, respectively) and postinfusional BMMCs (BMMC|Post). e, For each cell type, the fraction of clonotype groups in CR and non-CR groups is shown. Only cell types with more than 100 cells are depicted. f, Comparison of TCR clonotype diversity between CR and non-CR. Statistical significance between the two groups was estimated using a two-sided Wilcoxon rank-sum test. The center of the box plots indicates the median, and the upper and lower bounds of the boxes indicate first and third interquartile ranges. Whiskers extend to 1.5× interquartile range. The following are the numbers of biologically independent samples for the contrasts: PBMC|Pre: n = 5 for CR and non-CR; PBMC|Post: n = 4 for CR and n = 5 for non-CR; BMMC|Post: n = 4 for CR and n = 2 for non-CR. g,h, Analyses of CAR transcripts in the top ten TCR clones from participants 12 (g) and 14 (h) showed parallel existence of transfected (CAR+ = yellow bars) and nontransfected (CAR– = red bars) T cells of the same clonotype. Examples shown are from participant P12 with two hyperexpanded Ide-cel+ CAR T cell clones (clone 1 and 2; g) and participant P14 with Ide-cel+ CAR T cells with low clonality (h). Longitudinal TCR tracking of the top ten clones at apheresis and at 30 days after reinfusion is depicted on the right in both g and h. Abbreviations: TCM, central memory T cells; TEM, effector memory T cells; TPEX, precursor exhausted T cells; TEX, exhausted T cells; MAIT, mucosal-associated invariant T cells; CTL EOMES, eomesodermin-expressing cytotoxic lymphocytes; CTL GNLY, granulysin-expressing cytotoxic lymphocytes; CTL Exh, exhausted cytotoxic lymphocytes; TFH, follicular helper T cells; TH17, interleukin-17 (IL-17)-producing helper T cells; Treg, regulatory T cells. NA, not applicable.
Next, we compared single-cell composition (Fig. 7a), transcriptomes (Fig. 7b) and proteomes of the T cell compartment and in silico extracted CAR T cells (Fig. 7c,d (Ide-cel) and Fig. 7e,f (Cilta-cel)) after infusion with preinfusional T cells to characterize functional changes caused by lentiviral transfection. In line with the TCR clonality analyses, the relative dominance of CD8+ TEMRA and effector memory T cells delineated responders from nonresponders at all time points (Fig. 7a and Extended Data Fig. 3a), whereas the number of naive CD8+ T cells decreased after treatment in individuals in CR (Extended Data Fig. 3a). Gene sets associated with immune cell activation were enriched in responders and downregulated in nonresponders (Fig. 7b and Extended Data Fig. 3b).

a, Differences in T cell-type composition between CR and non-CR pre- and postinfusional PBMCs and BMMCs. For each cell type, the log fold change in mean cell fraction between non-CR and CR samples was calculated. The cell fraction calculation includes all cell types (the denominator is the sum of all cells analyzed). Significance was estimated using speckle. For clarity, only cell types with a fold change of >1.5 are shown. b, GO term enrichment analysis of DEGs (non-CR versus CR) for BMMCs and PBMCs. c,e, After in silico extraction of CAR T cells from peripheral blood, DEGs between CAR T cells and preinfusional T cells were compared. Because we saw marked differences in TCR clonality in participant P12 (c) who showed two hyperexpanded CAR+ T cell clones compared to low to intermediate clones in other individuals treated with Ide-cel (participant P14; e), two separate analyses to identify DEGs were performed. CAR T cells exhibited gene expression profiles associated with cytotoxicity and exhaustion. Downregulated gene sets mapped to T cell differentiation and activation in Ide-cel-treated individuals (Extended Data Fig. 4). d,f, Phenotyping based on single-cell proteomes (d and f, n = 61 surface antigens) showed significant expression (FDR < 0.05 and fold change of <1.5 or fold change of >1.5) of antigens associated with exhaustion and senescence in hyperexpanded Ide-cel+ CAR T cells from participant P12 (for example, CD57/B3GAT1, CD38, killer cell lectin-like receptor subfamily G member 1 (KLRG1) and CD279/PD1), whereas markers of activation or naive-like T cell state were downregulated (d). By contrast, Ide-cel+ CAR T cells with low to intermediate clonality (participant P14; f) were characterized by decreased cell surface expression of inhibitory molecules such as T cell immunoglobulin mucin receptor 3 (CD366/TIM-3) and KLRG1.
Compared to preinfusional CD4+ and CD8+ T cells, Ide-cel+ and Cilta-cel+ CAR T cells expressed significantly higher levels of genes associated with cytolysis (GZMA, GZMB and GNLY; Fig. 7c,e). Pathway analyses revealed a downregulation of pathways associated with T cell activation, cytotoxicity and chemotaxis in CD4+ and CD8+ CAR T cells (Extended Data Figs. 4 and 5). T cells ordered by inferred pseudotimes showed trajectories from T cells collected at apheresis over postinfusional, non-CAR T cells to CAR+ T cells alongside recurrently increased expression of cytolytic enzymes (Supplementary Figs. 1–8).
After analyzing single CAR T cell transcriptomes, we investigated surface protein expression. Hyperexpanded CAR T cells exhibited a more exhausted and senescent phenotype as indicated by higher levels of immune checkpoint proteins and NK cell receptors (PD1, CD38, B3GAT1/CD57 and KLRD1/CD94) as well as lower levels of markers associated with activation, such as CD11b, CD33 and CD69 and interleukin-7 receptor (IL-7R), which is linked to a naive-like T cell state20 (Fig. 7d). By contrast, nonhyperexpanded CAR T cell clonotypes (<10% of the entire TCR repertoire) showed a surface protein expression profile characterized by the downregulation of exhaustion/senescence markers (for example, TIM-3/CD366, KLRG1 and CD337; Fig. 7f) and higher expression levels of activation markers that were downregulated in hyperexpanded CAR T cell clones (Fig. 7d,f). Remarkably, the comparison of CAR T cells with nontransfected T cells after treatment revealed no significant differences in surface protein expression.
Although we were able to detect malignant circulating plasma cells at leukapheresis in responders and nonresponders (Fig. 8a), we hypothesized that the majority of CAR T cells are activated by bone marrow plasma cells. Therefore, we compared surface antigen expression of CAR T cells isolated from bone marrow aspirates to surface antigen expression of CAR T cells isolated from peripheral blood. CAR T cells from the bone marrow were characterized by higher expression levels of LeuM1/CD15 and lower levels of PD1, CD38 and B3GAT1/CD57 (Extended Data Fig. 5b).

a–c, Analysis of normalized surface expression of antigens on circulating malignant plasma cells (PCs) that are druggable with currently approved therapeutics (BCMA (a), CD38 (b) and SLAM family member 7 (SLAMF7; c)) before (n = 6 individuals with detectable circulating plasma cells; left) and after (n = 4 with detectable circulating plasma cells; right) CAR T cell infusion. Responders (CR; blue) had higher BCMA levels at baseline than nonresponders (non-CR; brown). There was no association between the expression of other targets and response. No loss of BCMA expression on circulating plasma cells was detected (a). Remarkably, resurfacing of CD38 after treatment with Ide-cel was observed, for example, in participant P10 (b), as a potential therapeutic option for relapsed disease. d,e, Longitudinal tracking of circulating malignant plasma cells (d) and T cell clones (e) during three lines of therapy in participant P1 (top, total number of cells; bottom, frequency). f, Based on genome-wide inferred CNV (losses, blue; gains, red), three different plasma cell clones were detected. The predominant clone at first relapse (clone 2, light blue, characterized by del13q) was susceptible to treatment with daratumumab/carfilzomib/dexamethasone (DKD), as indicated by a significant reduction of malignant circulating plasma cells 4 days after the start of therapy (d 4). After six cycles of DKD, the individual relapsed, and clone 1 (dark blue, characterized by the absence of del13q) gained dominance. Clone 1 was susceptible to treatment with elotuzumab/pomalidomide/dexamethasone (Elo-PD). At apheresis, the individual was in remission, as indicated by a significant reduction in circulating plasma cells. At this time point, clone 2 had regained dominance. Treatment with Ide-cel repressed clone 2 but had no therapeutic effect on clone 1. Clone 3 (orange, characterized by additional deletion of chromosome 19) remained on a subclonal level at all times. Besides the observed tiding of malignant plasma cell clones, we also tracked the coevolution of TCR repertoires (e). Treatment with Elo-PD resulted in a significant reduction in T cells at apheresis. A reduction in the number of CD8+ T cells at apheresis was associated with adverse outcome in our analysis. Relative abundances were plotted on the y axis for the five most abundant T cell clones.
Tiding and coevolution of malignant cells and T cells
After investigating the immune microenvironment before and after CAR T cell infusion and dissecting CAR T cell function in vitro and at single-cell resolution, we aimed at studying the compartment of malignant plasma cells to understand modes of resistance.
Sequencing single circulating plasma cells at apheresis and after CAR T cell infusion allowed the identification of surface antigens that are druggable with currently approved therapeutics (Fig. 8a–c). Responders showed higher levels of surface BCMA expression than nonresponders (Fig. 8a). No correlation between BCMA expression and response was detected for other druggable antigens, including CD38 (Fig. 8b) and SLAMF7 (Fig. 8c). In nonresponders, circulating malignant plasma cells were also detected after CAR T cell infusion. No loss of BCMA expression on plasma cells was detected (Fig. 8a). Resurfacing of CD38 expression after relapse from BCMA CAR T cells was detected in nonresponders (for example, participant P10 in Fig. 8b). Longitudinal tracking demonstrated clonal tiding and coevolution of malignant plasma cells and T cells (Fig. 8d,e). Based on inferred copy number variations (CNVs) from single-cell transcriptomes, we were able to identify daratumumab-resistant clones characterized by deletion of 13q (del13q; Fig. 8f) that could be addressed with elotuzumab/pomalidomide/dexamethasone treatment but resurged after Ide-cel treatment. Longitudinal TCR tracking showed expansion of T cells during the first 4 days of daratumumab/carfilzomib/dexamethasone treatment but diminished T cell receptor richness caused by elotuzumab/pomalidomide/dexamethasone treatment before apheresis (Fig. 8e). Although manufacturing was successful (437 × 106 CAR T cells per kg body weight infused, not out of specification; Supplementary Table 1), the participant showed early progression into secondary plasma cell leukemia within the first 100 days. Based on the detected CD38 positivity (Extended Data Fig. 6) and the presence of t(11;14), treatment with isatuximab/venetoclax/bortezomib/dexamethasone was initiated and resulted in a reduction in circulating plasma cells.
Discussion
CAR T cell therapies have led to unprecedented rates of deep and long-lasting remissions in heavily pretreated individuals with triple-class-exposed MM. However, most individuals relapse within 2 years after treatment, especially if residual disease can be detected in the months following CAR T cell infusion2,3,4,21. In our current study, we used single-cell multiomics to identify markers associated with response and resistance to CAR T cell therapy in MM.
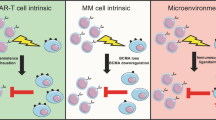
Immunoparesis is a cornerstone in the pathogenesis of MM and is pronounced in RRMM22,23,24. We found that an immunosuppressive microenvironment at apheresis characterized by monocytes expressing inhibitory immune checkpoints as well as numerically reduced and functionally impaired CD8+ T cells and NK cells were associated with adverse outcome after CAR T cell therapy. The expression of genes encoding PIM kinases was recurrently upregulated in NK cells, monocytes and dendritic cells. PIM kinases promote immune evasion and have been targeted in RRMM25,26. Combining PIM inhibition with anti-PD1 lead to long-term disease control by tumor-reactive T cells in a mouse model of melanoma, highlighting the potential of targeting PIM kinases to improve CAR T cell therapies27. Furthermore, we identified CD39 on monocytes as a potential target to modulate host immunity before and after CAR T cell therapy28. Avoiding exposure to lymphotoxic substances like bendamustine might be another strategy to avert an immunosuppressive microenvironment at apheresis29. These results demonstrate that individuals with poor outcome after anti-BCMA CAR T cell therapies can be identified before manufacturing and are characterized by an impaired immune system at apheresis. Choosing the right bridging therapy and combining it with agents that modulate immunosuppressive capabilities, especially of monocytes, might pave the way for successful treatment.
After identifying decreased T and NK cell activity in future nonresponders at apheresis, we hypothesized that CAR T cells manufactured from the respective impaired immune microenvironment are not functional compared to those from responders. However, in vitro analyses of CAR T cell cytotoxicity showed no significant differences between responders and nonresponders. This supports the theory that CAR T cells are functional outside a nonpermissive microenvironment, and modulating host immunity after CAR T cell infusion might contribute to increased efficacy of cellular immunotherapies.
Another finding that emphasizes the role of bystander non-CAR T cells was the expansion and activation of CD4+ and CD8+ non-CAR T cells in responders. Stimulation of bystander cells has been described after treatment with anti-CD19 CAR T cells30 and in a study of an academic anti-BCMA CAR T cell product in RRMM31. In line with this finding, we also found that hyperexpanded CAR T cell clones after therapy exhibited a terminally differentiated phenotype characterized by the expression of co-inhibitory markers such as PD1 (ref. 31). In line with our findings, the authors described a correlation between TCR diversity prior treatment and outcome.
In addition to hyperexpanded CAR T cell clones that were characterized by an exhausted and senescent phenotype, we characterized CAR T cells with low to intermediate clonality. Although the respective CD8+ CAR T cells also expressed a cytotoxic phenotype, they showed lower expression levels of markers associated with exhaustion (for example, TIM-3 and PD1). NK cell receptors like KLRG1/CD166 on cytotoxic T cells have been successfully targeted to improve T cell immunity32. Furthermore, the respective CAR T cells showed higher levels of activation markers like CD69 that were associated with improved outcome in the aforementioned study31. Unlike in other hematological malignancies, where CAR T cells can be detected up to decades after therapy and are applied in a curative manner33, CAR T cell persistence is limited to 6–12 months after infusion in MM2,21. Our results support the postinfusional use of inhibitors of immune checkpoints that were expressed on exhausted CAR T cells to prolong their activity. Because we were able to collect paired peripheral blood and bone marrow samples following treatment, we compared CAR T cells isolated from the two compartments. CAR T cells from the bone marrow had a more activated and less senescent phenotype than those from the peripheral blood, emphasizing that CAR T cell functionality depends on the compartment that they act in. This might have negative consequences for individuals with plasma cell leukemia and/or EMD, which becomes more prevalent after relapse from CAR T cell therapy34.
Last, we analyzed the compartment of malignant plasma cells. Biallelic loss of the TNFRSF17 (BCMA) locus on chromosome 16 has been described as a possible mode of resistance to anti-BCMA CAR T cells in RRMM5,6. This mechanism occurs only in a subset of individuals and was not observed in our cohort despite the detection of pre-existent del16p. However, responders had higher baseline levels of surface BCMA expression than nonresponders. In line with this observation, a recent preclinical study found that increasing BCMA surface density by augmenting its expression with all-trans-retinoic acid and preventing its shedding through γ-secretase inhibition improves efficacy of anti-BCMA CAR T cells35. Longitudinal single-cell sequencing of peripheral blood allowed a detailed characterization of tiding malignant plasma cell clones and T cells. In the past, we were able to demonstrate a negative impact of cytogenetically defined subclones in asymptomatic and newly diagnosed disease36,37. In our current analysis, we connected the existence of subclones to response and resistance to antibody-based triplet therapies and CAR T cells. The identification of druggable antigens before and after CAR T cell therapy might support therapeutic decision-making to optimize bridging therapy and treatment of relapse after CAR T cell infusion. Furthermore, analyzing the coevolution of the T cell compartment might identify the right moment to collect T cells for manufacturing to reduce the risk of out of specification productions.
Taken together, we identified an immunosuppressive microenvironment at the time of apheresis that is connected to adverse outcome after treatment with approved anti-BCMA CAR T cells for RRMM. Our single-cell multiomic analysis identified potential targets on non-CAR T cells, CAR T cells and malignant plasma cells to optimize the activity and durability of CAR T cells for future studies.
Methods
Treatment and sample collection
Individuals with RRMM were treated with commercial CAR T cell products (Ide-cel (Abecma) or Cilta-cel (CARVYKTI)) at the Department of Hematology, Cellular Therapy and Hemostaseology of the University Hospital in Leipzig, Germany. Individuals were treated in an inpatient setting, and lymphodepletion was performed according to the KarMMa2 and CARTITUDE-1 (refs. 3,4) studies that lead to the approval of the respective agents. Data and sample collection was approved by the local ethics committee (Leipzig Medical Biobank/LMB 311-15-24082015 and LMB-UCCL-2022_06) and was performed in accordance with the Declaration of Helsinki after obtaining written informed consent. The researchers were blinded to the conditions of the participants during data collection as well as sample collection and preparation. Peripheral blood was collected on the day of leukapheresis, the day of CAR T cell infusion (day 0) and days 7, 14, 30 and 100 after treatment. Bone marrow biopsies were performed on day 30 after CAR T cell therapy. Participants were followed up in our outpatient clinic, and responses were assessed according to International Myeloma Working Group recommendations38. For single-cell sequencing, BMMCs and peripheral blood PBMCs were isolated following standardized operating procedures using Leucosep tubes (Greiner Bio One) with a Ficoll gradient (Ficoll-Paque PLUS, VWR Avantor). Cells were frozen using a CoolCell LX container (Corning) and stored in the vapor phase of liquid nitrogen in an Askion HS200S freezer (Askion). For flow cytometry and cell culture experiments, fresh samples were handled as described below.
Single-cell RNA, TCR and BCR sequencing and surface proteomics
For single-cell multiomic analyses, PBMCs and BMMCs collected on the day of leukapheresis and approximately 30 days after CAR T cell infusion were thawed and assessed for quality by acridine orange and propidium iodide staining using automated cell counting. Before single-cell staining and library construction, samples with initial viability of <80% were subjected to dead cell removal using Viahance from BioPAL, Inc., following the manufacturer’s instructions.
A custom panel of 61 oligonucleotide-coupled antibodies (Singleron Biotechnologies) directed against cell surface proteins was used for surface proteome analyses (Supplementary Table 3). The oligonucleotides consisted of a linker to the antibody, a PCR handle, an antibody-specific barcode and a poly(A) tail. The antibody–oligonucleotide conjugate was then incubated with the cells of interest. After single-cell droplet encapsulation, the antibody-bound oligonucleotides act as synthetic transcripts and were then processed with 10x Genomics 5′ v2 chemistry to generate libraries for RNA-seq, BCR sequencing, TCR sequencing and surface protein expression. Libraries were sequenced on an Illumina NovaSeq using an S4 flowcell and v1.5 PE150 chemistry, targeting a mean coverage of 40,000 reads per cell for gene expression and 5,000 reads per cell for protein expression, TCR sequencing and BCR sequencing libraries.
Preprocessing of single-cell sequencing data
The CAR sequences for Ide-cel39 and Cilta-cel40 were obtained from the respective patents. Two separate custom references for Cilta-cal and Ide-cel have been created by adding the corresponding CAR sequences to the reference genome (GRCh38, Ensembl release 98) and GENCODE v32 annotation using the mkref function from CellRanger v7.1.0. The CellRanger multipipeline was used to process the single-cell gene expression, cell surface protein and V(D)J libraries. The feature reference .csv file, declaring antibody capture constructs and associated barcodes, is provided in Supplementary Table 3. The reference dataset required for the V(D)J contigs was provided by 10x Genomics (GRCh38 Human V(D)J Reference 7.1.0 (7 December 2022)).
For all subsequent analyses, R v4.2.2 was used. The R package scDblFinder v1.12.0 (ref. 41) was used to detect and remove barcodes that contained potential doublets. In addition, cell barcodes that met any of the following criteria were excluded: (1) fewer than 250 genes, (2) more than 8,000 genes, (3) fewer than 500 unique molecular identifiers (4) more than 100,000 unique molecular identifiers or (5) mitochondrial transcript fraction of more than 15%. The numbers of cells for each sample before and after the filter steps are listed in Supplementary Table 2. Raw gene expression data were normalized using the NormalizeData function (normalization.method = ‘LogNormalize’, scale.factor = 10,000) in Seurat v4.3.0 (ref. 18). Raw cell surface expression data were normalized by the centered log ratio method using the NormalizeData function (normalization.method = ‘CLR’ and margin = 2).
Annotation of cell identities and clonotyping
We tested SingleR42, multimodal reference mapping with Seurat18 and Mapcell43 for unsupervised annotation of cell types and found that the multimodal reference mapping approach has the best concordance with results from the TCR and BCR assay. We used PBMC and bone marrow44 reference datasets to annotate the PBMC and bone marrow samples, respectively. Seurat’s ‘Multimodal Reference Mapping’ vignette was used for label transfer from the reference to the query datasets (https://satijalab.org/seurat/articles/multimodal_reference_mapping.html). Label transfer consists of two main steps. First, the transfer anchors are identified with the FindTransferAnchors function. For this purpose, the query datasets were dimensionally reduced by applying the supervised principal component analysis (PCA) transformation calculated with the reference datasets. The first 50 dimensions were then used to define the neighbor search space. Second, these anchors were then used to transfer the cluster labels to the query dataset using the TransferData function, which additionally estimated a cell prediction score reflecting predictions that were supported by multiple consistent anchors. Only cells with a cell prediction score of >0.5 were retained for downstream analysis. Cells labeled as erythrocytes, erythroid progenitor cells and platelets were excluded from subsequent analysis. The reference datasets used include a coarse annotation of the T cell identities. To cover more subtypes, we reannotated T cells using the R package ProjecTILs v3.0.3 (ref. 45). First, we filtered for T cells using the R package scGate46, which requires as input (1) a normalized gene expression matrix and (2) a ‘gating model’ consisting of gene sets representing the cell population of interest. The gating model for CD4+ and CD8+ T cells was retrieved using the get_scGateDB() function. The T cell purification step was performed for each sample. To classify CD4+ and CD8+ T cell subtypes, we projected the T cells separately into two reference maps. As reference, we used the single-cell atlases for CD4+ and CD8+ T cells, which are part of the ProjecTILs framework (CD8: https://figshare.com/ndownloader/files/38921366; CD4: https://figshare.com/ndownloader/files/39012395). Running the ProjecTILs.classifier() function for each sample on the two maps allowed us to combine the identity predictions for a complete annotation of the T cell subtypes. Only cells that were unequivocally assigned to one cell subtype were annotated; the remaining cells (including those that received a label by more than one reference) were assigned to ‘Not Estimable’.
In addition, we annotated CD4+ and CD8+ T cell lineages based on the gene expression data using the following strategy. Given the raw counts, one cell was considered CD8+ or CD8– if the count value of CD8A or CD8B was >0 or ≤0, respectively. One cell was considered CD4+ or CD4– if the count value of CD4 was >0 or ≤0, respectively. This resulted in a classification of CD8–CD4+, CD8+CD4–, CD8+CD4+ and CD8–CD4– T cells. Double-positive cells (CD8+CD4+) classified by ProjecTILs as CD4+ or CD8+ were annotated as ‘Not Estimable’. We are aware that this is a rather conservative step; however, it allows us to avoid false-positive annotations. CD8+CD4+ cells classified as CD4+ in ProjecTILs were also labeled as ‘Not Estimable’ (and vice versa). To identify proliferating cell clusters, we applied the run_gsea function implemented in the clustifyr R package47 using previously published G2/M and S phase gene sets48 as queries. As suggested by the authors, an overclustering was performed. For this purpose, the cluster resolution was set to 1. For gene set enrichment, 1,000 permutations were performed. One cluster was significantly (P < 0.1) enriched with cell cycle genes from the G2/M and S phases and was annotated as cyclic accordingly. For T cell subtype markers, see Extended Data Fig. 7.
V(D)J data were analyzed using scRepertoire (v.1.11.0)49. Clonotypes were determined using the combineTCR and combineBCR functions. A TCR clonotype was defined as the combination of genes comprising the VDJC gene sequence and the nucleotide sequence of the CDR3 region (gene + nucleotide) for paired TCRα and TCRβ chains. BCR clonotypes were based on the V gene and >85% normalized Levenshtein distance of the nucleotide sequence.
Integration and UMAP visualizations
Integration and clustering analysis were performed using Seurat v4.3.0 (ref. 18). For each sample, the 2,000 most variable genes, excluding mitochondrial genes, were estimated using the ‘vst’ method of the FindVariableFeatures function. We then used the SelectIntegrationFeatures function to select 1,000 genes that were repeatedly shown to be highly variable in multiple samples. Thereafter, the data were standardized with ScaleData(), and a PCA was performed on the standardized expression data of the variable genes using the function RunPCA. We used 25 principal components, which explained ~90% of the variance, to integrate all samples using the Harmony50 method (v1.1) with the Seurat wrapper function RunHarmony with additional parameters (dim.use = 1:25, group.by.vars = c(‘SAMPLE_ID’)). Using the Harmony-corrected cell embeddings, we performed clustering by computing a shared nearest neighbors graph, as implemented in FindNeighbors(reduction = ‘harmony’, dims = 1:25). Cluster identification using the shared nearest neighbors graph was computed with the FindCluster function (Louvain algorithm). The same procedure was used for the ADT data but with the following differences: we used all surface proteins for PCA and 15 components for integration.
By analyzing transcriptomics and ADT data together, a WNN graph was determined with Seurat’s FindMultiModalNeighbors() function to represent a weighted combination of both modalities. The first 25 and 15 dimensions of the harmony-corrected RNA and ADT reductions, respectively, were used to create the graph. Subsequently, a UMAP embedding was constructed using the WNN graph.
Differential gene expression and enrichment analysis
Testing for differential gene expression was performed using the function FindMarkers implemented in Seurat. Genes with an FDR of <0.05 (Benjamini–Hochberg correction) and fold change of <1.5 or >1.5 were considered DEGs that were then used for GO enrichment analysis with the clusterProfiler package v.4.6.2 (ref. 51). In addition, we used the simplify function of clusterProfiler to remove redundant terms. GO terms with an FDR of <0.05 (as determined by the Benjamini–Hochberg procedure) were considered significant. Significant GO terms were ranked by rich factor, which is the number of DEGs in the term divided by the number of background genes in that term. In addition, a z score was calculated, which is the number of DEGs with a log fold change of >0 minus the number of DEGs with a log fold change of <0 divided by the square root of the number of term-associated genes.
Single-cell CNV inference
InferCNV52 was run to produce a heat map of CNVs in malignant plasma cells, with nonmalignant plasma cells as the reference. Malignant plasma cells were identified as cells forming a dominant clonotype constituting more than 90% of the plasma cells at any time point. Samples obtained from earlier time points where individuals were treated with daratumumab were also included in the analysis. Nonmalignant plasma cells were obtained from a publicly available dataset44. The output heat map showed significant variations of CNV signals in chromosomes 13 and 19, so hierarchical clustering was performed on CNV signals of only these two chromosomes to produce three CNV clusters. InferCNV was run again on these clusters separately with Markov chain Monte Carlo enabled to estimate CNV probabilities. Finally, a cell cluster was considered to harbor CNV for a particular locus if the CNV probability was more than 0.95 or if another cluster had a CNV probability of more than 0.95 while this cluster had a probability of more than 0.8 for that locus.
Cell–cell communication
Ligand–receptor signaling was inferred with iTalk53 using default settings. LIANA v0.1.12 (ref. 54) was used as a reference database consisting of manually curated resources in the context of cell–cell communication. All DEGs (non-CR versus CR) in preinfusional PBMCs were used as input.
Pseudotime trajectory analysis
Monocle 3 (ref. 55) was used for plotting pseudotime trajectories. To meet the format requirement of Monocle 3, Seurat objects were converted as cell datasets using the SeuratWrappers R package. Trajectories were estimated using the standard Monocle 3 workflow, which includes normalization, clustering, dimensionality reduction, visualization and differential expression. Trajectories were learned using the default parameter ‘learn_graph’ function. The node in the trajectory with the largest number of memory/naive-like T cells with the earliest time point in the vicinity was assigned pseudotime 0. Moran’s I-test was used to identify genes that vary with trajectory.
Immunophenotyping by flow cytometry
Blood samples from individuals with MM taken before CAR T cell treatment (day 0) and 7, 14, 30 and 100 days after treatment were analyzed by flow cytometry, as described previously56. For analysis of CAR immune status, biotin-labeled BCMA CAR Detection Reagent (2 µl; Miltenyi Biotec) was added directly to 100 µl of fresh EDTA-anticoagulated blood and incubated for 5 min at room temperature. After the addition of 2 ml of PBS and one washing step (500g, 5 min, room temperature), cell pellets were resuspended in 100 µl of PBS, and the following antibodies were added: 2 µl of anti-biotin-APC (Miltenyi Biotec) and 1 µl of CD4-BV786; 2.5 µl (each) of CD45-V500, CD8-APC-H7, CD19-APC-R700 and CD38-BV421; 5 µl (each) of CD3-PerCP, CD16-PE-Cy7, CD56-PE-Cy7, PD1-PE and HLA-DR-BV605 and 10 µl of CD14-FITC (all from Becton Dickinson). To analyze T cell differentiation, the following antibodies were added directly to 100 µl of EDTA-anticoagulated blood: 1 µl of CD4-BV786; 2.5 µl (each) of CD45-V500, CD8-APC-H7 and CD45RO-PE-Cy7; 5 µl (each) of CD3-PerCP, CD45RA-FITC and CD197-APC and 20 µl of CD31-PE (all from Becton Dickinson).
After incubation for 15 min at room temperature, red blood cells were lysed by the addition of 2 ml of lyse solution (Becton Dickinson) for 10 min. The cells were centrifuged (500g, 5 min, room temperature), and cell pellets were resuspended, washed with 2 ml of PBS (500g, 5 min, room temperature) and analyzed using a FACSLyric flow cytometer (Becton Dickinson). Data analysis was performed using the FACSSuite software (Becton Dickinson; see Supplementary Fig. 9 for the gating strategy).
Cell separation and culture
PBMC samples from individuals with MM (EDTA-anticoagulated blood) taken before CAR T cell treatment at day 0 and 7 days thereafter were isolated by either LymphoPrep (Progen) or Ficoll-Paque Plus (GE Healthcare) density centrifugation. PBMCs, resuspended in X-Vivo medium (Lonza) supplemented with 200 U ml–1 IL-2 (Peprotech) and 2% AB-Sera (Sigma-Aldrich), were transferred to culture plates coated with anti-CD3 (5 µg ml–1; Invitrogen) and co-stimulated with anti-CD28 (2 µg ml–1; Invitrogen) for up to 3 weeks to induce T cell proliferation. The MM cell line U266 was maintained in RPMI-1640 medium (Gibco) supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich). Culture expansion and medium changes were performed twice a week for T cells and U266 cells.
Detection of CAR expression in cell culture
Cultured (CAR) T cells (100,000) were washed twice in 100 µl of PBS + 2% FCS (400g, 2 min, 4 °C) before the addition of 1 µl of biotin-labeled BCMA CAR Detection Reagent (Miltenyi Biotec) to 50 µl of PBS + 2% FCS. After 10 min, two further washing steps in PBS + 2% FCS (400g, 2 min, 4 °C) were performed. Subsequently, 2.5 µl of anti-biotin-PE (Miltenyi Biotec) and 2.5 µl of anti-CD3-V450 (Becton Dickinson) in 50 µl of PBS + 2% FCS were added for 10 min. Two additional washing steps were performed (100 µl of PBS + 2% FCS; 400g, 2 min, 4 °C), and cells were resuspended in 100 µl of PBS + 2% FCS before the addition of 5 µl of viability dye 7-AAD (Becton Dickinson) for 10 min. Samples were diluted with 200 µl of PBS + 2% FCS, and cells were immediately analyzed using a FACSCanto II flow cytometer (Becton Dickinson). Data analysis was performed using FlowJo 10.8.1 (FlowJo).
Cytotoxicity assay
The target cell line U266 (1 × 106 cells per ml) was labeled with 1 µM VPD450 (Beckton Dickinson) and incubated for 15 min at 37 °C. After three washing steps in PBS, cells were resuspended (1 × 106 cells per ml) in X-Vivo medium (Lonza) supplemented with 2% AB-Serum (Sigma-Aldrich) and were used for coculture with effector cells.
The expanded (CAR) T cells (effector cells) at day 0 before CAR T cell treatment and 7 days thereafter were collected and resuspended (1 × 106 cells per ml) in X-Vivo medium (Lonza) supplemented with 2% AB-Serum (Sigma-Aldrich). Subsequently, 200,000 effector cells (T cells from days 0 and 7) and 30,000 target cells (U266) were cocultured in two 96-well U-bottom plates. U266 target cells (30,000) cultured without effector cells served as controls.
Cell were either incubated for 24 h at 37 °C and 5% CO2 or were placed on ice and analyzed immediately to avoid unwanted target cell killing. To quantify the number of cells, 5,000 flow cytometry count beads (Invitrogen) were added to each well.
Cells were washed twice with 100 µl of PBS (400g, 2 min, 4 °C) before Fixable Viability Dye FVD eFlour 780 (1:500; Thermo Fisher Scientific) was added for 15 min at 4 °C. After three additional washing steps in PBS + 2% FCS (100 µl), 2.5 µl of anti-CD3-FITC (Biolegend) in 50 µl of PBS + 2% FCS was added for 10 min at 4 °C. Cells were washed twice in PBS + 2% FCS (400g, 2 min, 4 °C), fixed with 1% formaldehyde and analyzed by a FACSCanto II flow cytometer (Becton Dickinson) by counting 2,500 bead events. Cells cocultured for 24 h or 0 h were treated the same way. Data analysis was performed using FlowJo 10.8.1 (FlowJo).
Statistics and reproducibility
Statistical analysis was performed in R v.4.2.2. No data distribution assumptions were applied to scRNA-seq data. Therefore, the comparisons between groups on the gene expression level were performed with a nonparametric approach (two-tailed Wilcoxon rank-sum test) that does not assume normally distributed data. For testing of differences in cell-type proportions, a normal distribution of the data was not assumed. Using the speckle R package, an arcsin square root transformation was performed. Significant differences were estimated using empirical Bayes moderated t-statistics (two sided) implemented in the speckle package. GO term enrichment was calculated using a hypergeometric test. Estimated probabilities of progression-free survival were calculated using the Kaplan–Meier method and the log-rank test to evaluate differences between survival distributions. All statistical tests are listed in the corresponding Methods sections and figure legends for clarity. No participants or data were excluded from the analyses because we included the first ten consecutive participants treated with CAR T cells at our institution. No randomization was performed, and the authors performing wet lab experiments and primary bioinformatic analyses were blinded to outcome.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq and scRNA-seq, BCR sequencing and TCR sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession code GSE234261. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
Processing and analysis code related to this study is deposited in the GitHub repository at https://github.com/fraunhofer-izi/Rade_Grieb_et_al_2023.
References
Merz, M. et al. Adjusted comparison of outcomes between patients from CARTITUDE-1 versus multiple Myeloma Patients with Prior Exposure to PI, Imid and Anti-CD-38 from a German Registry. Cancers 13, 5996 (2021).
Munshi, N. C. et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 384, 705–716 (2021).
Berdeja, J. G. et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 398, 314–324 (2021).
Martin, T. et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J. Clin. Oncol. 41, 1265–1274 (2022).
Da Vià, M. C. et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 27, 616–619 (2021).
Samur, M. K. et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 12, 868 (2021).
Gagelmann, N. et al. Access to and affordability of CAR T-cell therapy in multiple myeloma: an EBMT position paper. Lancet Haematol. 9, e786–e795 (2022).
Gazeau, N. et al. Effective anti-BCMA retreatment in multiple myeloma. Blood Adv. 5, 3016–3020 (2021).
Deng, H. et al. Efficacy of humanized anti-BCMA CAR T cell therapy in relapsed/refractory multiple myeloma patients with and without extramedullary disease. Front. Immunol. 12, 720571 (2021).
Haradhvala, N. J. et al. Distinct cellular dynamics associated with response to CAR-T therapy for refractory B cell lymphoma. Nat. Med. 28, 1848–1859 (2022).
Sheih, A. et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 11, 219 (2020).
Bai, Z. et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci. Adv. 8, eabj2820 (2022).
Boiarsky, R. et al. Single cell characterization of myeloma and its precursor conditions reveals transcriptional signatures of early tumorigenesis. Nat. Commun. 13, 7040 (2022).
Cohen, Y. C. et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 27, 491–503 (2021).
Dutta, A. K. et al. Single-cell profiling of tumour evolution in multiple myeloma—opportunities for precision medicine. Nat. Rev. Clin. Oncol. 19, 223–236 (2022).
Ledergor, G. et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat. Med. 24, 1867–1876 (2018).
Merz, M. et al. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat. Commun. 13, 807 (2022).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Phipson, B. et al. propeller: testing for differences in cell type proportions in single cell data. Bioinformatics 38, 4720–4726 (2022).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020).
Cohen, A. D. et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 129, 2210–2221 (2019).
Nakamura, K., Smyth, M. J. & Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 136, 2731–2740 (2020).
Merz, M. et al. Spatiotemporal assessment of immunogenomic heterogeneity in multiple myeloma. Blood Adv. 7, 718–733 (2022).
Zavidij, O. et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 1, 493–506 (2020).
Clements, A. N. & Warfel, N. A. Targeting PIM kinases to improve the efficacy of immunotherapy. Cells 11, 3700 (2022).
Keane, N. A., Reidy, M., Natoni, A., Raab, M. S. & O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 5, e325 (2015).
Chatterjee, S. et al. Targeting PIM kinase with PD1 inhibition improves immunotherapeutic antitumor T-cell response. Clin. Cancer Res. 25, 1036–1049 (2019).
Moesta, A. K., Li, X.-Y. & Smyth, M. J. Targeting CD39 in cancer. Nat. Rev. Immunol. 20, 739–755 (2020).
Vucinic, V. et al. S287: factors influencing autologous lymphocyte collections for chimeric antigen receptor (CAR) T-cells—the role of T-cell senescence. HemaSphere 6, 188 (2022).
Chen, P.-H. et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 5, e134612 (2020).
Dhodapkar, K. M. et al. Changes in bone marrow tumor and immune cells correlate with durability of remissions following BCMA CAR T therapy in myeloma. Blood Cancer Discov. 3, 490–501 (2022).
Mathewson, N. D. et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298 (2021).
Melenhorst, J. J. et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 602, 503–509 (2022).
Wang, B. et al. Chimeric antigen receptor T cell therapy in the relapsed or refractory multiple myeloma with extramedullary disease—a single institution observation in China. Blood 136, 6 (2020).
García-Guerrero, E. et al. All-trans retinoic acid works synergistically with the γ-secretase inhibitor crenigacestat to augment BCMA on multiple myeloma and the efficacy of BCMA-CAR T cells. Haematologica 108, 568–580 (2023).
Merz, M. et al. Cytogenetic subclone formation and evolution in progressive smoldering multiple myeloma. Leukemia 34, 1192–1196 (2020).
Merz, M. et al. Prognostic significance of cytogenetic heterogeneity in patients with newly diagnosed multiple myeloma. Blood Adv. 2, 1–9 (2017).
Kumar, S. et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 17, e328–e346 (2016).
Campbell, T. et al. Uses of anti-BCMA chimeric antigen receptors. Worldwide patent WO2021091978A1 (2021).
Schecter, J. M. & Fan, X. BCMA-targeted CAR-T cell therapy for multiple myeloma. Worldwide patent WO2022116086A1 (2022).
Germain, P.-L., Lun, A., Meixide, C. G., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979 (2022).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172 (2019).
Koh, W. & Hoon, S. MapCell: learning a comparative cell type distance metric with Siamese neural nets with applications toward cell-type identification across experimental datasets. Front. Cell Dev. Biol. 9, 767897 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Andreatta, M. et al. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 12, 2965 (2021).
Andreatta, M., Berenstein, A. J. & Carmona, S. J. scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets. Bioinformatics 38, 2642–2644 (2022).
Fu, R. et al. clustifyr: an R package for automated single-cell RNA sequencing cluster classification. F1000Res. 9, 223 (2020).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Borcherding, N., Bormann, N. L. & Kraus, G. scRepertoire: an R-based toolkit for single-cell immune receptor analysis. F1000Res. 9, 47 (2020).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Tickle, T., Tirosh, I., Brown, M. & Haas, B. InferCNV: inferring copy number alterations from tumor single cell RNA-seq data. GitHub https://github.com/broadinstitute/inferCNV/wiki (2023).
Wang, Y. et al. iTALK: an R package to characterize and illustrate intercellular communication. Preprint at bioRxiv https://doi.org/10.1101/507871 (2019).
Dimitrov, D. et al. Comparison of methods and resources for cell–cell communication inference from single-cell RNA-seq data. Nat. Commun. 13, 3224 (2022).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Blache, U. et al. Advanced flow cytometry assays for immune monitoring of CAR-T cell applications. Front. Immunol. 12, 658314 (2021).
Acknowledgements
We thank S. Scharf from the Leipzig Medical Biobank for handling cryopreserved samples and D. Bretschneider, C. Mueller, K. Wildenberger and C. Wilhelm for performing fluorescence in situ hybridization analyses on clinical samples. We express our gratitude toward the participants and their families who participated in this analysis. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SPP µbone) and EU HORIZON Project CERTAINTY (101136379), and M.M. received financial support from grants from the International Myeloma Society, SpringWorks and Janssen.
Author information
Authors and Affiliations
Contributions
Conception and design: M.R., N.G., R.W., V.V. and M.M. Acquisition of data (acquired and managed participants, provided facilities, fluorescence in situ hybridization, flow cytometry, biobanking, in vitro studies and so on): L.F., P.B., A.B., S.F., P.F., C.K., S. Heyn, A.S.K., R.B., S.Y.W., E.B., S. Hoffmann, J.U., B.S., S. Hell, M.J., S.S., K.H.M., G.-N.F., M.H., U.S., K.R., U.K., U.P., V.V. and M.M. Analysis and interpretation of data (for example, statistical analysis, biostatistics and computational analysis): M.R., N.G., R.W., J. Sia, S.X., L.F., J. Scolnick, L.V., K.R., V. V. and M.M. Writing, review and/or revision of the manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
S.F.: consultant and/or speaker fees from Novartis Pharma, Janssen-Cilag, Vertex Pharmaceuticals (Germany), Kite/Gilead Sciences, MSGO and Bristol-Myers Squibb. U.K.: consultant and/or speaker fees from AstraZeneca, Affimed, Glycostem, GammaDelta, Zelluna, Miltenyi Biotec and Novartis Pharma and Bristol-Myers Squibb. M.M.: advisory boards/honoraria/research support from Amgen, BMS, Celgene, Gilead, Janssen, Stemline, Springworks and Takeda. K.H.M.: BMS (consultancy and honoraria), AbbVie (honoraria, research funding), Pfizer (honoraria), Otsuka (honoraria), Janssen (honoraria) and Novartis (consultancy). U.P.: Syros (consultancy, honoraria, research funding), MDS Foundation (membership on an entity’s Board of Directors or advisory committees), Silence Therapeutics (consultancy, honoraria, research funding), Celgene (honoraria), Takeda (consultancy, honoraria, research funding), Fibrogen (research funding), Servier (consultancy, honoraria, research funding), Roche (research funding), Merck (research funding), Amgen (consultancy, research funding), Novartis (consultancy, honoraria, research funding), AbbVie (consultancy), Curis (consultancy, research funding), Janssen Biotech (consultancy, research funding), Jazz (consultancy, honoraria, research funding), BeiGene (research funding), Geron (consultancy, research funding) and Bristol-Myers Squibb (consultancy, honoraria, membership on an entity’s Board of Directors or advisory committees, other, travel support, medical writing support, research funding). M.J.: Novartis (honoraria), Amgen (honoraria), Pfizer (honoraria), Blueprint Medicine (honoraria), BMS (honoraria) and Jazz (honoraria). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cancer thanks Marco Davila and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Enrichment analysis for post-infusional BMMCs and PBMCs.
GO term enrichment analysis of significantly differentially expressed genes (non-responders vs. responders) for BMMCs and PBMCs. Terms are ranked by rich factor, which is the number of DE genes in the term divided by the number of background genes in that term. Dot plot depict the highest ranked significantly enriched GO terms for biological processes (FDR < 0.05). The dot size indicates the z-score, which is the number of DE genes with logFC >0 minus the number of DE genes with logFC <0 divided by the square root of the number of term-associated genes. Grey/white dots indicate the same number of genes with a logFC >0 and <0. Cell type abbreviations: NK = natural killer; Mono = monocyte; cDC = classical dendritic cell.
Extended Data Fig. 2 Comparing post- and pre-infusional BMMCs and PBMCs in nonCR and CR.
a, Comparison between post- and pre-infusional cell types in PBMCs from responders (CR) and non-responders (nonCR). The number of biologically independent samples for the contrasts is as follows. CR: n = 4 for post- and n = 5 for pre-infusional groups. nonCR: n = 5 for post- and n = 5 for pre-infusional groups. Significant differences (unadjusted p-values) were estimated using the empirical Bayes moderated t-statistics (two-sided) implemented in the speckle package (* p = 0.1, ** p = 0,05, *** p = 0.01, **** p = 0.001, p < 0.0001 = *****). The exact p-values for CR are as follows: NK (p = 0.00210), NK_CD56bright (p = 0.01849), B memory (p = 0.08501) and pDC (p = 0,02643), B intermediate (p = 0.074792) and HSPC (p = 0,09701). The exact p-value for nonCR, is as follows: CD14 Mono (p = 0.03526). The center of the box plots indicates the median, and the upper and lower bounds of the boxes indicate first and third interquartile ranges. Whiskers extends to 1.5 x interquartile range. b, Differential gene expression analysis comparing post- with pre-infusional BMMCs and PBMCs in CR and nonCR Only cell types with significant genes are shown (FDR < 0.05 and fold change <1.5 or >1.5). A positive log fold change indicates upregulation in post-infusional samples. c, GO term enrichment analysis of significantly differentially expressed genes (post- vs. pre-infusional). Terms are ranked by rich factor, which is the number of DE genes in the term divided by the number of background genes in that term. Dot plots depict the highest ranked significantly enriched GO terms for biological processes (FDR < 0.05). The dot size indicates the z-score, which is the number of DE genes with logFC >0 minus the number of DE genes with logFC <0 divided by the square root of the number of term-associated genes. Grey/white dots indicate the same number of genes with a logFC >0 and <0. Cell type abbreviations: NK = natural killer; gdT-Cell = gamma delta T cell; Mono = monocyte; cDC = classical dendritic cell; pDC = plasmacytoid dendritic cell; Cilta cel = Ciltacabtagene autoleucel; Ide cel = Idecabtagene vicleucel; GMP = granulocyte-monocyte progenitor, HSPC = Hematopoietic stem and progenitor cell.
Extended Data Fig. 3 Comparing post- with pre-infusional T cell subtypes in nonCR and CR.
a, Differences in T cell subtype composition between post- and pre-infusional PBMCs from CR and nonCR. For each cell type, the log fold change in mean cell fraction between post- and pre-infusional samples was calculated with the R package speckle based on arcsin square root transformation. The cell fraction calculation includes all cell types (the denominator is the sum of all cells analyzed). For clarity, only cell types with a fold change >1.5 are shown. b, GO term enrichment analysis for DEGs comparing post- with pre-infusional PBMCs in CR and nonCR. GO terms are ranked by rich factor, which is the number of DE genes in the term divided by the number of background genes in that term. Dot plot depict the highest ranked significantly enriched GO terms for biological processes (FDR < 0.05). Cell type abbreviations: CM = central memory; EM = effector memory; TEMRA = effector memory cells re-expressing CD45RA; TPEX = precursor exhausted T cells; TEX = exhausted T cell; MAIT = Mucosal-associated invariant T cell; CTL_EOMES = eomesodermin expressing cytotoxic lymphocytes; CTL_GNLY = granulysin expressing cytotoxic lymphocytes; CTL_Exh = exhausted cytotoxic lymphocytes; Tfh T follicular helper cells; Treg = regulatory T cells; gdT = gamma delta T cell.
Extended Data Fig. 4 Enrichment analysis in CAR T cells (CD4 and CD8) compared to pre-infusional T cells from patients treated with Ide-cel.
GO term enrichment analysis of significantly differentially expressed genes (CAR vs. pre-infusional T cells) in participants 12 (a) and 14 (b). Terms are ranked by rich factor, which is the number of DE genes in the term divided by the number of background genes in that term. Dot plot depict the highest ranked significantly enriched GO terms for biological processes (FDR < 0.05).
Extended Data Fig. 5 Enrichment analysis in CAR T cells (T cell subtypes) compared to pre-infusional T cells from patients treated with Ide-cel.
a, GO term enrichment analysis of significantly differentially expressed genes (CAR vs. pre-infusional T cells) in PBMCs for patient 12. No significantly enriched GO terms were found for patient 14. b, Differential cell surface protein expression for CD4 and CD8 T-cells when comparing CAR+ from PBMCs with CAR+ from BMMCs for patient 12 (BMMCs CD4 n = 41; CD8 430 // PBMCs CD4 n = 214; CD8 4699). Genes with an FDR < 0.05 and fold change <1.5 or >1.5 were considered as significantly differentially expressed. Cell type abbreviations: CM = central memory; EM = effector memory; TEMRA = effector memory cells re-expressing CD45RA; TEX = exhausted T cell.
Extended Data Fig. 6 Analysis of CD38 surface protein expression in patient 01 with samples available after exposure/becoming refractory to a CD38 antibody as well as after discontinuing the antibody.
Patient 01 had progressed with a plasma cell leukemia after frontline treatment with 4 cycles bortezomib/cyclophosphamide/dexamethasone (VCD), high-dose melphalan and autologous transplantation (HDM + ASCT) and lenalidomide (Len) maintenance. Circulating malignant plasma cells (n = 7071) showed homogeneous CD38 protein expression, which was significantly decreased 4 days after starting Daratumumab/Carfilzomib/Dexamethasone (Dara/Carf/Dex). The patient relapsed 9 months later. CD38 expression on circulating malignant plasma cells significantly increased after 4 days of 3rd line treatment with Elotuzumab/Pomalidomide/Dexamethasone (Elo/Pom/Dex). CD38 expression levels increased further upon relapsed from treatment with Idecabtagene vicleucel (Ide-cel) 2 months after the last dose of Daratumumab.
Extended Data Fig. 7 Marker genes for T cell identities.
DE genes for each T cell identity were determined using the Wilcoxon rank-sum test. Only CAR negative cells were analyzed. Genes with an FDR < 0.05 and an absolute fold change >1.5 were considered statistically significant. Genes are ranked according to FDR. For each cell identity, 5 significant genes are shown. The color intensity indicates the standardized average expression level in a cell identity. The dot size indicates the percentage of expressing cells within each cell identity of the corresponding genes.
Supplementary information
Supplementary Information
Supplementary Figs. 1–8 (pseudotime trajectory analysis of preinfusional and postinfusional CD4+CD8+ T cells and CAR T cells) and Supplementary Fig. 9 (gating strategy).
Supplementary Tables
Supplementary Table 1. Summary of ten individuals treated with Ide-cel or Cilta-cel (participants P7 and P8). Except for participants P8 and P10, all individuals had at least one high-risk cytogenetic feature detected by fluorescence in situ hybridization (mostly chromosome 1 abnormalities). Bridging therapy consisted mainly of antibody-based three-drug combinations. Participant P9 was treated with high-dose chemotherapy and autologous stem cell transplantation before CAR T cell infusion. Abbreviations: m, male; f, female; FISH, fluorescence in situ hybridization; Elo/Pom/Dex, elotuzumab/pomalidomid/dexamethasone; Benda/Carf/Dex, bendamustine/carfilzomib/dexamethasone; Dara/Pom/Dex, daratumumab/pomalidomide/dexamethasone; HD-Melphalan, high-dose melphalan; ASCT, autologous stem cell transplantation; Benda/Dex, bendamustine/dexamethasone; IsaPomDex, isatuximab/pomalidomide/dexamethasone; Dara/Len/Dex, daratumumab/lenalidomide/dexamethasone; Isa/Carf/Dex, isatuximab/carfilzomib/dexamethasone. Supplementary Table 2. Summary of samples and quality control data from scRNA-seq, TCR and BCR sequencing and surface proteomics. Abbreviations: PR, partial remission; PD, progressive disease; UMI, unique molecular identifier. Supplementary Table 3. Feature reference .csv file declaring antibody capture constructs and associated barcodes. The CellRanger multipipeline requires this configuration .csv file as input. Supplementary Table 4. Summary of differential gene expression analyses. Testing for differential gene expression was performed using the function FindMarkers (Wilcoxon rank-sum test) implemented in Seurat. Genes with an FDR of <0.05 (Benjamini–Hochberg correction) and fold change of <1.5 or >1.5 were considered DEGs. Summaries include comparisons of different cell types between nonresponders and responders from PBMCs after CAR T cell therapy (post-PBMC; non-CR versus CR), BMMCs after CAR T cell therapy (post-BMMC; non-CR versus CR), PBMCs collected at apheresis (pre-PBMC; non-CR versus CR) and comparisons of PBMCs after CAR T cell therapy versus PBMCs at apheresis in responders (CR; post versus pre) and nonresponders (non-CR; post versus pre). Supplementary Table 5. Summary of differential gene expression analyses for T cell identities. Testing for differential gene expression was performed using the function FindMarkers (Wilcoxon rank-sum test) implemented in Seurat. Genes with an FDR of <0.05 (Benjamini–Hochberg correction) and fold change of <1.5 or >1.5 were considered DEGs. Summaries include comparisons of T cell identities between nonresponders and responders from PBMCs at apheresis (pre-PB; T cell; non-CR versus CR), PBMCs after CAR T cell therapy (post-PB; T cell; non-CR versus CR), BMMCs after CAR T cell therapy (post-BM; T cell; non-CR versus CR) and comparisons of PBMCs after CAR T cell therapy versus PBMCs at apheresis in responders (CR; PB; T cell; post versus pre) and nonresponders (non-CR; PB; T cell post versus pre), CAR T cells versus preinfusional T cells in participant P12 (CAR versus pre-T cell) and CAR T cells versus preinfusional T cells in participant P14 (CAR versus pre-T cell).
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rade, M., Grieb, N., Weiss, R. et al. Single-cell multiomic dissection of response and resistance to chimeric antigen receptor T cells against BCMA in relapsed multiple myeloma. Nat Cancer (2024). https://doi.org/10.1038/s43018-024-00763-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43018-024-00763-8
