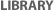Figure 1
Figure 1












« Prev Next »
Although most healthy adults who contract the flu experience relatively minor symptoms, influenza and other respiratory viruses are a serious health threat to the U.S. population at large. Children and the elderly are particularly susceptible to the flu epidemic that sweeps the country each winter. Moreover, pandemics involving influenza strains to which most people do not have immunity occur periodically, attended by high levels of mortality and disastrous consequences to public health. Properly preparing for the influenza threat is thus a constant challenge for public health officials, especially given the fact that the viruses that cause infection can mutate rapidly and reassort to form new strains, and that these viruses also have the ability to reside in multiple animal hosts. Unfortunately, despite intensive research over the course of decades, much remains unknown about why some influenza strains are highly transmissible and why certain flu viruses cause such severe disease.
Flu Statistics
In the United States, seasonal influenza epidemics typically claim the lives of about 30,000 people each year and cause hospitalization of more than 100,000 (Reid & Tautenberger, 2003). Every two or three years, more virulent strains circulate, increasing death tolls by approximately 10,000 to 15,000 individuals. These seasonal epidemics are the result of antigenic drift, a phenomenon caused by mutations in two key viral genes due to an error-prone RNA polymerase.
Less frequently, however, new and particularly virulent strains of influenza arise, which cause worldwide pandemics that are accompanied by greatly increased death tolls. These strains occur because of the phenomenon known as antigenic shift, in which humans are infected with avian influenza viruses or viruses that contain a combination of genes from human and avian sources. Since 1900, three of these pandemics have occurred. The first, which took place in 1918 and was referred to as "Spanish" influenza, was the deadliest, claiming an estimated 40 million lives worldwide in less than a year (Palese, 2004). Unlike weaker flu strains that are more of a threat to the elderly, this flu claimed the lives of many young people, including children and young adults. In fact, people under age 65 accounted for 99% of the deaths attributed to this strain, whereas subsequent pandemics claimed many fewer people from this age group. Later epidemics occurred in 1957, when the "Asian" flu killed 70,000 people in the United States, and in 1968, when the "Hong Kong" strain killed 30,000 Americans (Reid & Tautenberger, 2003).
The Influenza Virus and Its Genome
The name "influenza" is derived from the Latin word for "influence," and the pathogens that cause this disease are RNA viruses from the family Orthomyxoviridae. The genomes of all influenza viruses are composed of eight single-stranded RNA segments (Figure 1). These RNAs are negative-sense molecules, meaning that they must be copied into positive-sense molecules in order to direct the production of proteins.
With the HA and NA genes, the influenza A genome contains eight genes encoding 11 proteins. These proteins include three RNA polymerases that function together as a complex required by the virus to replicate its RNA genome. Interestingly, these polymerases have been shown to have high error rates due to a lack of proofreading ability, which leads to high mutation rates in replicated viral genomes and therefore rapid rates of viral evolution. This high rate of mutation and evolution is one source of influenza virus genetic diversity. The influenza genome also encodes additional structural proteins necessary to form the capsid, the nucleoprotein (NP), and the proteins NS1 (nonstructural protein 1) and NS2/nuclear export protein (NEP), whose roles are still being investigated. Still other proteins encoded by the viral genome include membrane proteins M1 and M2 (which are needed for nuclear export and several other functions) and, of course, HA and NA (which play roles in viral attachment and release from host cells, respectively).
There are three basic types of influenza viruses: A, B, and C. Influenza B and C viruses only infect humans, so novel antigens are not introduced from other species. Only influenza A viruses infect nonhuman hosts, and a reassortment of genes can occur between those subtypes that typically infect animals and those that infect humans, resulting in antigenic shift and potential pandemics. Epidemics of seasonal influenza occur due to influenza A or B viruses.
As in all viruses, the genome of an influenza virus particle is encased in a capsid that consists of protein. The influenza A capsid (Figure 2) contains the antigenic glycoproteins hemagglutinin (HA) and neuraminidase (NA); several hundred molecules of each protein are needed to form the capsid. These proteins are the parts of the virus that are recognized as foreign by a host's immune system, thus eliciting an immune response. Because many different subtypes of the influenza A hemagglutinin and neuraminidase proteins exist, the human immune system is frequently challenged with new antigens. For example, point mutations in the HA and NA genes can lead to changes in antigenicity that allow a virus to infect people who were either infected or vaccinated with a previously circulating virus. This phenomenon is referred to as antigenic drift. In addition to humans, other animals can be infected with or serve as a reservoir for influenza, and outbreaks have been seen in poultry, pigs, horses, seals, and camels (Hayden & Palese, 1997). When a strain is named, the host (if not human), the location where the virus originated, the strain number, the year of isolation, and the HA/NA subtype are all included in the name.
Figure 2: Electron micrograph of influenza A virus particles.
The genome of influenza A viruses consists of eight single-stranded RNA segments, and the viral particle has two major glycoproteins on its surface: hemagglutinin and neuraminidase.
Figure courtesy of M-T. Hsu and P. Palese, Mount Sinai School of Medicine, New York, New York. All rights reserved. 
Due to the segmented nature of the influenza genome, in which coding sequences are located on individual RNA strands, genomes are readily shuffled in host cells that are infected with more than one flu virus. For example, when a cell is infected with influenza viruses from different species, reassortment can result in progeny viruses that contain genes from strains that normally infect birds and genes from strains that normally infect humans, leading to the creation of new strains that have never been seen in most hosts. Moreover, because at least 16 different hemagglutinin subtypes and nine different neuraminidase subtypes have been characterized, many different combinations of capsid proteins are possible. Of these subtypes, three subtypes of hemagglutinin (H1, H2, and H3) and two subtypes of neuraminidase (N1 and N2) have caused sustained epidemics in the human population. Birds are hosts for all influenza A subtypes and are the reservoir from which new HA subtypes are introduced into humans (Palese, 2004).
Deciphering the 1918 Epidemic
Because influenza viruses were not isolated and cultured until the 1930s, it was not possible to study the origin of the 1918 Spanish flu pandemic at the time of this virus's outbreak; indeed, the virus was not extensively studied until the last decade of the twentieth century. Specifically, in 1997, both frozen and formalin-fixed lung tissue from Spanish flu victims was used to extract nucleic acid and sequence the 1918 influenza genome (Tautenberger et al., 1997). The samples were derived from a U.S. soldier who died in New York, another U.S. solider who died in South Carolina, an Inuit woman who died in Alaska, and two victims from the Royal London Hospital in the United Kingdom. These strains shared a sequence identity of 99%.
The sequence of the 1918 influenza genome proved to be puzzling and did not immediately answer researchers' questions regarding the strain's origin. In the later flu pandemics that occurred in 1957 and 1968, the responsible strains appeared to have arisen through reassortment of avian-derived HA genes into human strains. In contrast, the HA gene of the 1918 strain is most closely related to an influenza isolate obtained from swine (Reid & Tautenberger, 2003). The 1918 HA sequence bears some similarities to those sequences commonly seen among avian strains, but it differs from them much more than the strains responsible for the later pandemics. Indeed, when comparing the HA genes from all three pandemic strains to those from both Eurasian and North American avian species, the 1918 HA genes bear the least resemblance. Furthermore, the later strains display fewer sequence differences overall and resemble Eurasian avian sequences much more closely than they do North American avian sequences (Table 1). They also resemble avian sequences more closely than they do any mammalian sequence. Taken together, these data suggest that pigs may have been an intermediate host for the 1918 strain, although this remains to be demonstrated.
| Pandemic Strain | Number of Differences from North American Avian Consensus Sequences | Number of Differences from Eurasian Avian Consensus Sequences |
| 1918 | 24 | 24 |
| 1957 | 19 | 5 |
| 1968 | 14 | 7 |
| Table 1: Number of amino acid differences between pandemic HA and avian subtype consensus sequences (Adapted from Reid & Tautenberger 2003) | ||
Underdiagnosis of Influenza and Implications for Public Health
Recent evidence indicates that many physicians frequently fail to diagnose influenza, or they do not specifically distinguish the flu from other respiratory viruses that can cause similar symptoms. For example, a 2006 study appearing in the New England Journal of Medicine concluded that "most influenza infections in children were not diagnosed clinically" (Poehling et al., 2006). As part of a project called the New Vaccine Surveillance Network, which was sponsored by the U.S. Centers for Disease Control, the researchers involved in this study tracked attending physicians' diagnoses for pediatric inpatients and outpatients reporting flu-like symptoms at several American hospitals. They also conducted their own independent diagnoses of the patients, including laboratory confirmation of pathogen presence. The investigators reported that only one-third of the hospitalized patients identified as influenza positive by the surveillance team had been tested for flu as part of the care they received. Moreover, of the children with surveillance-confirmed infections, only 28% of inpatients and 17% of outpatients had received clinical diagnoses of influenza. The remaining patients were given diagnoses of various other conditions, including asthma, pneumonia, or a "nonspecific diagnosis of viral infection," when in fact they were influenza positive. The study authors thus concluded that "surveillance that relies on data from physician-directed testing alone substantially underestimates the influenza burden," reflecting a "lack of recognition of influenza during most visits." Clearly, a lack of sufficient diagnosis and tracking makes development of effective preventative strategies and vaccines difficult.
Although the basic biology and genetics of influenza viruses are fairly well understood, heading off future pandemics requires a better understanding of past pandemics and the factors that contribute to virulence, as well as a thorough commitment to tracking the viruses that are circulating in the population by way of focused public health efforts.
References and Recommended Reading
Hayden, F., & Palese, P. Influenza virus. In Clinical Virology, ed. D. D. Richman (New York, Churchill Livingston, 1997), 891–920
Horimoto, T., et al. Influenza: Lessons from past pandemics, warnings from current incidents. Nature Reviews Microbiology 3, 591–600 (2005) doi:10.1038/nrmicro1208 (link to article)
Nelson, M. I., et al. The evolution of epidemic influenza. Nature Reviews Genetics 8, 196–205 (2007) doi:10.1038/nrg2053 (link to article)
Palese, P. Influenza: Old and new threats. Nature Medicine 10, S82–S87 (2004) (link to article)
Poehling, K. A., et al. The underrecognized burden of influenza in young children. New England Journal of Medicine 355, 31–40 (2006)
Reid, A. H., & Tautenberger, J. K. The origin of the 1918 pandemic influenza virus: A continuing enigma. Journal of General Virology 84, 2285–2292 (2003)
Tautenberger, J. K., et al. Initial genetic characterization of the 1918 "Spanish" influenza virus. Science 275, 1793–1796 (1997) doi:10.1126/science.275.5307.1793