
Abstract
Molecular mechanisms which underpin compound leaf development in some legumes have been reported, but there is no previous study on the molecular genetic control of compound leaf formation in Vigna unguiculata (cowpea), an important dryland legume of African origin. In most studied species with compound leaves, class 1 KNOTTED-LIKE HOMEOBOX genes expressed in developing leaf primordia sustain morphogenetic activity, allowing leaf dissection and the development of leaflets. Other genes, such as, SINGLE LEAFLET1 in Medicago truncatula and Trifoliate in Solanum lycopersicum, are also implicated in regulating compound leaf patterning. To set the pace for an in-depth understanding of the genetics of compound leaf development in cowpea, we applied RNA-seq and whole genome shotgun sequence datasets of a spontaneous cowpea unifoliate mutant and its trifoliate wild-type cultivar to conduct comparative reference-based gene expression, de novo genome-wide isoform switch, and genome variant analyses between the two genotypes. Our results suggest that genomic variants upstream of LATE ELONGATED HYPOCOTYL and down-stream of REVEILLE4, BRASSINOSTERIOD INSENSITIVE1 and LATERAL ORGAN BOUNDARIES result in down-regulation of key components of cowpea circadian rhythm central oscillator and brassinosteroid signaling, resulting in unifoliate leaves and brassinosteroid-deficient-like phenotypes. We have stated hypotheses that will guide follow-up studies expected to provide more insights.
Similar content being viewed by others


Introduction
Morphogenetic variation between simple and compound leaf forms begin with the developing leaf primordia on the peripheral zone of shoot apical meristem (SAM), and the expression of class 1 KNOTTED-LIKE HOMEOBOX (KNOXI) genes in developing leaf primordia is thought to play a defining role in this variation1,2,3,4. Unraveling the molecular mechanisms controlling the differences in simple and compound leaf morphogenesis has a long history and has remained relevant, following the flexibility in the molecular events that result in diverse leaf patterns5,6,7,8,9. The phases of leaf ontogenesis—initiation, primary morphogenesis, and secondary morphogenesis—are similar in simple and compound leaves2,10,11,12. However, the formation of separated leaflet primordia in species with compound leaves, based on which leaflet number is determined, distinguishes compound and simple leaf primary morphogenesis3. The distinguishing pattern of expression of the KNOXI genes in tomato, in comparison with KNOXI expressions in species with simple leaves, established the molecular basis for differentiating the development of simple and compound leaves9. Efforts to investigate the mechanisms of compound leaf development in legumes have implicated other genes in the regulation of compound leaf patterning. While KNOX1 genes are absent in the developing leaf primordia of Medicago truncatula and Pisum sativum, legumes of the inverted repeat-lacking clade (IRLC), legume-specific FLORICAULA/LEAFY orthologs, SINGLE LEAFLET1 and UNIFOLIATA, respectively, regulate compound leaf development in the two legume species13,14. Conversely, SHOOT MERISTEMLESS-like KNOX genes are associated with compound leaf development in Vigna radiata15, suggesting that absence of KNOXI genes expression in leaf primordia of some legumes species may be restricted to legumes in the IRLC15,16,17,18. In soybean, the legume-specific transcription factor, E1, earlier reported to regulate flowering and maturation19,20,21, is also associated with leaf development22; however, there is no evidence which associates E1 with compound leaf formation in soybean.
Cowpea (Vigna unguiculata; 2n = 2x = 22) is an economically important legume crop23,24, which produces two opposite simple leaves as the first pair of leaves, and subsequently bears alternate trifoliate compound leaves. Genetic inheritance of unifoliate mutant loci in a natural unifoliate mutant25 and unifoliate mutant segregants in F2 and advanced generations of crosses involving wild-type trifoliate cowpea cultivars26 have been studied, but the underlying molecular mechanisms of cowpea leaf development has not been previously reported. Here, using shotgun sequences and RNA-seq datasets of a spontaneous unifoliate cowpea mutant and its wild-type variety, we combined reference-based differential gene expression, de novo genome-wide isoform switch (IS), and genome variant analyses to identify phenotype-causing candidate genes and significantly enriched or depleted Gene Ontology (GO) terms associated with the mutant. Our study suggests that nucleotide variations upstream of LATE ELONGATED HYPOCOTYL (LHY) and down-stream of REVEILLE4 (RVE4), BRASSINOSTEROID INSENSITIVE 1 (BRI1) and LATERAL ORGAN BOUNDARIES (LOB) repress the components of cowpea circadian rhythm central oscillator and brassinosteroid (BR) signaling, resulting in the development of unifoliate leaves and phenotypes associated with BR deficiency27,28. This serves as a foundation for further research towards elucidating the genetic control of compound leaf patterning in cowpea.
Results
Genetic characterization of a novel natural dominant mutant in cowpea
We identified a spontaneous mutant bearing subsessile dark green unifoliate curled leaves (UCL) with elongated petiole-like pulvini (Fig. 1a, b) in the 2021 cowpea experimental field of Arid Land Research Center (ALRC), Tottori University, Japan. The mutant arose as a spontaneous segregant from the self-pollinated progeny of a cowpea wild-type variety, IT86D-1010 (Fig. 1a). Compared with the wild-type variety, the mutant plants (heterozygotes and homozygotes) are smaller, bear more structurally deformed than normal flowers (Fig. 1a,c), and yield significantly (p < 0.01) less shoot biomass and pods (Table 1). Production of abnormal flowers by cowpea unifoliate mutants is not unusual25,26, but the underlying molecular mechanism is still unclear. The low pod yield is largely caused by the obvious reduced self-pollination resulting from the structural deformity of the flowers, especially the protrusion and physical separation of stamens from the pistil of some flowers (Fig. 1c). Our observation of six self-pollinated generations of the mutant (heterozygote, homozygote and revertant) revealed that the mutant phenotype consistently expresses in both heterozygotes and homozygotes, with segregation in the progenies of heterozygotes conforming to the expected Mendelian 1:2:1 ratio for complete dominance; no segregation was observed in the progenies of homozygous mutant and revertant plants (Table 2). Also, reciprocal crosses between the mutant plants and three wild-type cowpea varieties (Sasaque, IT97K-499–35 and IT86D-1010) confirmed that the mutant locus is dominant. Crosses involving homozygous mutant plants (UCLHom) and the wild-type cowpea varieties produced only heterozygous mutant plants (UCLHet), while the progenies of crosses between UCLHet plants and the wild-type cowpea varieties segregated in approximately 1:1 ratio, UCLHet:Revertant (Table S1).

Morphological variation between cowpea wild-type (IT86D-1010) and UCL mutant. (a) Wild-type, UCLHet and UCLHom plants; (b) Wild-type and UCL mutant leaves; c. Wild-type and UCL mutant flowers; UCL, unifoliate curled leaf, UCLHet, UCL heterozygous mutant; UCLHom, UCL homozygous mutant.
Sequencing quality and reads mapping metrics
We conducted two categories of RNA-seq-based differential gene expression analyses between UCLHom and IT86D-1010: reference-based differential gene expression analysis based on normalized gene counts, and genome-wide differential isoform usage (DIU) analysis, using de novo assembled transcripts. As the mutant allele is dominant and the phenotypic difference between UCLHet and UCLHom is obvious (Fig. 1a), UCLHom plants used for preparation of RNA-seq libraries were selected from confirmed non-segregating self-reproducing progeny of UCLHom (Table 2). Strand-specific paired-end poly(A)-selected RNA-seq libraries of two bulked biological replicates, composed of 20 plants each of the purified RNA samples of UCLHom and IT86D-1010 were prepared using TruSeq Stranded mRNA Library Prep Kit (Illumina). The libraries were sequenced with DNBSEQ-G400 (MGISEQ-2000RS) short reads sequencer (Data S1 – S8). Initial read quality assessment was done with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), after which the sequences were processed by fastp29. Both FastQC and fastp results indicated sequencing adequacy (See Figure S1a and S1b; Table S2). The fastp results showed 10.7 to 13 Gb/library, 109 – 133 M reads/library, and average read length of 100 b (See Table S2). RNA STAR mapper30 was applied to align the fastp-filtered sequences to Vunguiculata_540_v1.0 reference genome, with Vunguiculata_540_v1.2 reference annotation as the gene model (https://data.jgi.doe.gov/refine-download/phytozome?genome_id=540). A multi-sample 2-pass mapping was conducted to enable accurate discovery and quantification of splice junctions31. Post-alignment RNA-seq-specific quality metrics aggregated with MultiQC32 confirmed the suitability of the datasets for the intended downstream applications. Importantly, in all the samples, over 80% of the reads were uniquely mapped, and more than 80% of the mapped reads mapped on CDS exons. Gene coverage plot showed no bias, and junction saturation plots showed stability of known splice junctions, indicating saturated sequencing depth (See Figure S2)33.
We used UCLHom, UCLHet and wild-type revertant F2 segregants of IT97K-499–35 × UCLHom hybrid to map candidate mutations in the UCL mutant genome. The genome sequences of the UCL mutant bulk (bulked DNA samples of 10 plants each of UCLHet and UCLHom) and wild-type revertant (bulked DNA samples of 20 revertant plants) generated by DNBSEQ-G400 short reads sequencer (Data S9 – S12) were analyzed by FastQC. The results, which showed phred scores of more than 30 (See Figure S1c and S1d), indicated sequencing adequacy. However, we still trimmed the sequences with fastp before applying Bowtie234 to align the short reads to Vunguiculata_540_v1.0 reference genome. The fastp-filtering results showed an average read length of 150 b in the two libraries, about 295.2 M sequence reads and 44 Gb in the revertant bulk library, and 365.8 M reads and 54.7 Gb in the UCL mutant bulk library (See Table S2).
Reference-based differential gene expression and functionally enriched or depleted GO terms in the UCL mutant genome
Differential gene expression between the wild-type and UCL mutant was analyzed using gene counts obtained by implementing featureCounts35 on the mapped collection of the datasets produced by RNA STAR mapper. With the aid of DESeq236, we normalized the gene counts and identified 2,106 significantly differentially expressed genes (DEGs) in the UCL mutant genome (adj. p < 0.05 and abs(log2(FC)) > 1; abs, absolute; FC, fold change). Of the DEGs, 1,068 (~ 51%) are up-regulated and 1,038 (~ 49%) are down-regulated (Fig. 2; see also Table S3). To make sense of the large number of the DEGs, we sorted the genes from the most highly expressed to the least expressed in the wild-type, and applied g:Profiler, a leading functional enrichment web resource, to identify significantly enriched or depleted functional Gene Ontology (GO) terms37,38. A multi-query functional enrichment analysis was performed, with each query consisting of 50 genes. The results of the functional enrichment analysis showed 21 significantly enriched terms, and 10 significantly depleted terms (p (SCS) < 0.05; SCS, Set Counts and Sizes)38 (See Table S4 and S5), indicating a complex interplay of biological functions involving plastid, chloroplast, cell wall and extracellular region in the UCL mutant genome. The identified enriched or depleted functional GO terms will guide further research on compound leaf patterning and other functionally compromised mechanisms in the mutant.

Heatmap of RNA-seq-based differential gene expression between wild-type (IT86D-101) and UCL homozygous mutant. UCL, unifoliate curled leaf; IT86D1_wildtype_bulk1, wild-type biological bulk replicate 1; IT86D2_wildtype_bulk2, wild-type biological bulk replicate 2; UCLHom1_mutant_bulk1, UCL homozygous mutant biological bulk replicate 1; UCLHom2_mutant_bulk2, UCL homozygous mutant biological bulk replicate 2.
Compound leaf and UCL mutant phenotype-associated DEGs
Of the functionally characterized genes associated with compound leaf development5, we identified, among the DEGs in the UCL mutant, cowpea orthologs (best protein matches) of SHOOT MERISTEMLESS (STM; Vigun06g157800), LATERAL ORGAN BOUNDARIES (LOB; Vigun02g150500), BRASSINOSTEROID INSENSITIVE 1 (BRI1; Vigun02g046500), and cytochrome P450 (CYP450) genes, including CONSTITUTIVE PHOTOMORPHOGENIC DWARF (CPD; Vigun01g226500) (Fig. 3a–d), which is highly expressed in leaves and strongly associated with BRI1-dependent BR signaling39.

Differential expressions of gene candidates associated with UCL mutant phenotype. (a) Up-regulation of VunSTM in UCLHom; (b) Up-regulation of VunLOB in UCLHom; (c) Up-regulation of VunCPD in UCLHom; (d) Down-regulation of BRI1 in UCLHom; e. Down-regulation of VunBES1/BZR1 in UCLHom; (f) Up-regulation of VunBKI1 in UCLHom; (g) Down-regulation of VunCCA1 in UCLHom; (h) Down-regulation of VunLHY in UCLHom; (i) Down-regulation of VunLNK in UCLHom; (j) Down-regulation of VunRVE1 in UCLHom; (k) Down-regulation of VunRVE4 in UCLHom; (l) Down-regulation of VunRVE7 in UCLHom; UCL, unifoliate curled leaf; UCLHom, UCL homozygous mutant.
The insignificant expression of cowpea STM (VunSTM) in the wild-type and its high (~ 4000-fold) expression in the UCL mutant (Fig. 3a) suggests that the expression of HOMEOBOX/KNOX-like genes in cowpea is suppressed under wild-type conditions, which is consistent with reports that indicated that KNOX-like genes are excluded from the leaves of some legumes1,5. M. truncatula ortholog of LOB, ELONGATED PETIOLULE1 (ELP1)/PETIOLULE-LIKE PULVINUS (PLP), is involved in leaf movements, mediated by rhythmic swelling, and shrinking of the motor cells of pulvinus40,41. Altered expression of ELP1/PLP produces mutant plants with elongated pulvini, where the motor cells are being replaced by petiole-like epidermal cells42. As shown in Fig. 1b, the pulvini of the leaves of the UCL mutant are elongated and curled. This suggests that the up-regulation of the VunLOB (Fig. 3b) in the UCL mutant results in petiolule-like pulvini (Fig. 1b), comparable to the response of M. truncatula to ectopic expression of ELP1/PLP40,42.
Also, LOB negatively regulates BR accumulation43, hence the differential expressions of CPD and BRI1 in the UCL mutant genome are particularly informative. CPD, known to be more actively transcribed than other BR-biosynthetic CYP450 genes, is one of the CYP450 genes that encode BRs biosynthesis rate-limiting enzymes44,45,46. Consequently, the up-regulation of CPD in the UCL mutant is likely associated with deficiency in BRs, as transcription of CPD is negatively controlled by BRs, and a brassinosteroid insensitive 1 (bri1) mutant has been reported to have an up-regulated expression of CPD in A. thaliana39,47,48. In A. thaliana, BRI1, a leucine-rich repeat receptor kinase (LRR-RLK), initiates BR signaling cascade, and BRI1 KINASE INHIBITOR1 (BKI1) prevents the signaling of BRI149,50. Among other components of BRs signaling cascade, BRI1-EMS SUPPRESSOR1 (BES1) and BRASSINOZOLE-RESISTANT1 (BZR1) are positive regulators of BRI51,52. Intriguingly, we found that cowpea orthologs of BRI1, BES1/BZR1 (Vigun05g181100) and BKI1 (Vigun09g239100) are differentially expressed in the UCL mutant: BRI1 and BES1/BZR1 are down-regulated, while BKI1 is up-regulated (Fig. 3d–f). Based on the expressions of the BR signaling-associated genes in the UCL mutant, we contend that the up-regulation of VunBKI1 in the UCL mutant negatively regulates the activity of VunBRI1 and VunBES1/BRZ1, resulting in BR deficiency, indicated by the BR-deficient-like phenotypes observed in the mutant (Fig. 1a, b)27,28.
Predicted expression and co-expression analyses associate CCA1, LHY, STM, CPD, RVE and LNK with leaf development
CIRCADIAN CLOCK ASSOCIATED 1 (CCA1; Vigun09g004100) and LATE ELONGATED HYPOCOTYL (LHY; Vigun10g153300) MYB-related TFs are also highly down-regulated in UCLHom (Fig. 3g, h). To investigate the association of these MYB TFs with leaf development in cowpea, we conducted in silico gene expression analysis for the TFs and the DEGs associated with compound leaf development. As we are not aware of any cowpea electronic gene expression browser, we used soybean (non-IRLC legume) orthologs of cowpea CCA1 (Glyma.03g261800), LHY (Glyma.07G048500), STM (Glyma.15G111900), and CPD (Glyma.11G228900) to predict the organs where the genes likely express during cowpea development. Using the Soybean electronic fluorescent pictograph (eFP) browser at http://bar.utoronto.ca53,54, under Developmental Map view, we found that CCA1, LHY, STM, and CPD show medium to strong expressions in SAM and leaves (See Figure S3), indicating their involvement in leaf development, especially LHY and STM which are strongly expressed in SAM. The expression of genes associated with compound leaf morphogenesis in SAM and young leaf primordia is required to maintain primary morphogenetic activity in compound leaf development5,55.
To identify other DEGs that are highly positively correlated with cowpea CCA1 and LHY (r ≥ 0.7), we used A. thaliana orthologs of CCA1 and LHY, At5g02840 and At1g01060, respectively, to conduct co-expression analysis at http://bar.utoronto.ca/ExpressionAngler/56. From our results, apart from CCA1 and LHY highly co-expressing with each other, a total of 41 other genes are highly co-expressed with either CCA1 or LHY or both (See Table S7). More importantly, among all the co-expressed genes, only seven genes are differentially expressed (down-regulated) in the mutant (See emboldened gene IDs in Table S7), indicating that the other co-expressed genes are not involved in the UCL mutation. Among the seven co-expressed DEGs are DENTIN SIALOPHOSPHOPROTEIN-LIKE PROTEIN (Vigun06g223900), orthologous to A. thaliana NIGHT LIGHT-INDUCIBLE AND CLOCK-REGULATED1 (LNK1) (Fig. 3i), and PROTEIN REIVELLE4-RELATED (RVE4; Vigun07g078900) (Fig. 3k), orthologous to A. thaliana REVEILLE8 (RVE8). LNK1 and RV8 are transcriptional coactivators and positive regulators of the components of circadian rhythm central oscillator; RVE8 specifically targets the evening element of CCA1/LHY promoters57,58,59. Considering the importance of RVE genes in circadian clock-regulated developmental processes in plants59,60,61,62, we further examined the expressions of other VunRVE gene homologs in the wild-type and UCL mutant genomes. We found that VunRVE1 and VunRVE7 are also down-regulated in the UCL mutant (Fig. 3j, l). This suggests that the components of the circadian clock central oscillator and their coactivators are repressed in the UCL mutant genome. Like CCA1, LHY, STM and CPD, which are strongly expressed in leaves and/or SAM, soybean orthologs of VunRVE1 (Glyma.10g048500), VunRVE4 (Glyma.15g053000), VunRVE7 (Glyma.18g044200) and VunLNK (Glyma.16g217700) are strongly expressed in leaves and/or SAM (See Figure S3e – S3h), suggesting their association with leaf development.
De novo transcriptome assembly and quantification
To ensure a comprehensive identification and quantification of all the transcripts necessary for a reliable analysis of alternative splicing, we applied the mapped sequences of the RNA-seq datasets to perform de novo transcriptome reconstruction. With the aid of StingTie63, we assembled and quantified the transcripts from the mapped collection of the libraries, guided by Vunguiculata_540_v1.0 reference genome and Vunguiculata_540_v1.2 reference annotation. To generate a new non-redundant reference transcriptome annotation, the assembled transcripts were merged with Vunguiculata_540_v1.2 reference annotation, using StingTie-merge63. We evaluated the annotation accuracy using GFFCompare64 to compare the merged reference annotation with Vunguiculata_540_v1.2 reference annotation. From the accuracy statistics output of GFFCompare (See Note S1), at all feature levels, sensitivity ranged from 99.1% to 100%, while precision ranged from 88.7% to 97.7%, indicating high proportions of query features that agree with corresponding reference annotation features. Also, out of the 61,283 query mRNAs in 33,697 loci across all the input datasets, no exon or locus was missed. While only 1 intron out of 150,963 introns (~ 0%) was missed, 3,921 novel exons, 2007 novel introns, and 2,043 novel loci were identified (See Note S1). Apart from validating the annotation accuracy, the GFFCompare statistics show that, although gene/transcript databases are large and reasonably representative, a de novo transcriptome assembly has the power of revealing novel transcript structures which are not represented in databases. The validated reference transcriptome we generated from StingTie-merge enabled us to accurately quantify transcript-level expressions, which we used to conduct genome-wide analysis of alternative splicing (AS) and isoform switch (IS).
Isoform switching underlies differential expressions of CCA1, LHY and CPD in the UCL mutant
To identify the predominant AS type resulting in significant functional switches from wild-type to mutant, the transcript-level gene expressions of all the transcripts in the RNA-seq datasets were analyzed using IsoformSwitchAnalyzeR65. We generated a raw switch list and their corresponding nucleotide and amino acid sequences, and used the amino acid and nucleotide sequences to, respectively, identify coding domains, and predict the coding potential of the transcripts, using CPAT66. With the switch list, protein domains and coding potential data, genome-wide AS and their functional consequences in the mutant were analyzed. The genome-wide gene expression plots indicate that there are genes in the mutant genome that are both differentially expressed (large log2FC) and contain isoform switches (red color) (See Figure S3a)65. The results of comparing the number of isoforms significantly differentially used between the wild-type and mutant genomes resulting in at least one splice event indicate that there is statistically significant alternative transcription termination sites (ATTS) events in the mutant genome (See Figure S3b)65. Also, consequence enrichment analysis shows that there is a significant gain in nonsense-mediated mRNA decay (NMD) insensitivity in the mutant genome (See Figure S3c). This implies a likely accumulation of truncated proteins translated from NMD-insensitive transcripts in the mutant67,68,69. NMD, a conserved mRNA quality control mechanism in eukaryotes, identifies and destroys aberrant mRNA containing premature termination codons (PTCs), and regulates the expression of normal transcripts67,68,70,71,72,73.
From the genome-wide AS analysis, we discovered 286 genes with statistically significant switches resulting in predicted functional consequences (q < 0.05) in the mutant genome, out of which 32 genes are significantly differentially expressed (See Table S6. The 32 IS-dependent DEGs are among the 2,106 DEGs identified using the reference-based approach of differential gene expression analysis, validating the two approaches of differential gene expression analysis we adopted. The two IS-dependent most down-regulated genes are CIRCADIAN CLOCK ASSOCIATED 1 (CCA1; Vigun09g004100) and LATE ELONGATED HYPOCOTYL (LHY; Vigun10g153300) MYB-related TFs (Fig. 4a and b), whose orthologs in A. thaliana are key components of circadian rhythm central oscillator74,75,76,77. Interestingly, we found that the up-regulation of CPD in the UCL mutant is also IS-dependent (Fig. 4c), which suggests a possible cross-talk between the expressions of the isoforms of CCA1 or LHY or both and CPD in cowpea.

Isoform switch-dependent differential expressions of genes associated with UCL mutant. (a) Down-regulation of VunCCA1 in UCLHom mutant resulting from decreased expression and usage of the wild-type isoform; (b) Down-regulation of VunLHY in UCLHom mutant resulting from decreased expressions and usage of wild-type isoforms; (c) Up-regulation of VunCPD resulting from higher expression and usage of the mutant isoform; UCL, unifoliate curled leaf; UCLHom, UCL homozygous mutant.
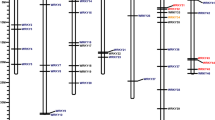
Genomic variants in the UCL mutant candidate genes
We applied FreeBayes, a reputable Bayesian genetic variant detector78, to jointly call a total of 7,700,700 variant sites from the UCL mutant and revertant mapped genome sequences. To identify UCL genome-specific variants, MiMohD VCF Filter79 was applied to extract 69,449 homozygous variant sites spread across all the chromosomes in the UCL mutant genome corresponding to homozygous wild-type loci in the revertant genome. This filtering ensured that recombinant variant sites which are common to UCL mutant and revertant genomes were excluded from the UCL variants list. The filtered variants were annotated using snpEff eff80, and reported with MiMohD Report Variants79. The variants are genome-wide and are mostly in the intergenic, upstream, and downstream regions (Fig. 5a), making the UCL mutant a potential source of genes for cowpea breeding. Of the 12 UCL phenotype-associated DEGs in the UCL mutant genome (Fig. 2), only LHY, RVE4, BRI1 and LOB have either upstream or downstream nucleotide variations in the UCL mutant genome (Fig. 5b–e; See also Table S8 – 11). LHY is affected by two upstream substitutions, TCG/CTA (chrVu10: 37,344,133) and CA/TC (chrVu10: 37,344,604) (See Table S8); a downstream TATATATGTA insertion (chrVu07: 10,891,928) affects RVE4 (See Table S9); three downstream substitutions, A/T (chrVu02: 18,476,009), C/T (chrVu02: 18,476,011) and ACA/GCC (chrVu02: 18,483,367), affect BRI1 (See Table S10), and LOB is affected by two downstream substitutions G/T (chrVu02: 29,738,970) and A/T (chrVu02: 29,738,992) (See Table S11). LHY and RVE4 co-express with CCA1 (See Table S7); LNK, RVE1 and RVE7 are positive regulators of CCA1/LHY57,58,59, and BRI1, CPD, BES1/BZR1, BKI1 and LOB are all associated with BR biosynthesis and accumulation43,49,50. The functional relationships among the gene candidates suggest that the differential expressions of the candidates without genomic variants in the UCL mutant are functional responses to expressions of LHY, RVE4, BRI1 and LOB.

Regions of genomic variants in UCL mutant. (a) Total genomic variants; (b) VunLHY variants; (c) VunRVE4 variants; (d) VunBRI1 variants; (e). VunLOB variants; UCL, unifoliate curled leaf.
Discussion
IS-dependent repression of circadian rhythm-regulated genes control leaf patterning in cowpea
For optimal utilization and allocation of resources, plants rely on endogenous circadian rhythms to synchronize internal physiological processes with diurnal cycles and seasonal environmental changes76,81,82. About 6% of gene expression patterns, and various developmental processes in plants exhibit circadian rhythms83,84. In A. thaliana, CCA1, LHY and TOC1 are components of the central oscillator of circadian clock; CCA1 and LHY, which are morning expressed genes85,86, are needed for the maintenance of circadian rhythm under constant light, and TOC1, an evening expressed gene87,88, expresses earlier than expected, when CCA1 and LHY are deactivated75. Disruptions in the expressions of CCA1 and LHY affect the period of circadian rhythms in gene expression and leaf movements57,58,75,85,89,90. RVE genes and their transcriptional coactivators, LNK1 and LNK2, are positive regulators of the components of central oscillator of circadian clock59,60,61,76. The roles of CCA1, LHY, TOC1 and other circadian rhythm-associated genes have also been studied in various legume species91. In cowpea, VunTOC1, VunLHY, VunELF3 and VunGI are associated with circadian clock function in seed filling and leaves92. Our results have shown that VunCCA1, VunLHY, VunLNK, VunRVE1, VunRVE4 and VunRVE7 are all down-regulated in the UCL mutant, suggesting a disruption in the mutant’s circadian clock central oscillator. While the down-regulation of VunCCA1 and VunLHY are IS-dependent, the repression of VunLNK and the three VunRVE genes are not associated with IS. Of all the circadian rhythm-regulated DEGs associated with leaf development, only VunLHY and one of its positive regulators, VunRVE4, have mutations in their upstream and downstream regions, respectively. This suggests that the repressed expressions of the other functionally related candidates are likely dependent on the repression of VunLHY or VunRVE4 or both. We, therefore, hypothesize that the genomic variants in VunLHY or VunRVE4 or both resulted in decreased expressions of the wild-type isoforms of VunLHY and VunCCA1 in the UCL mutant, compelling the compensatory usage of the alternative isoforms that resulted in the development of unifoliate leaves in the mutant. Put differently, optimal expressions of the wild-type isoforms of VunLHY and VunCCA1, and their positive regulators, VunLNK, VunRVE1, VunRVE4 and VunRVE7, are required for compound leaf development in cowpea. Previous studies have associated S. lycopersicum and M. truncatula MYB TFs with compound leaf patterning55,93,94,95,96. Increased expression of the tomato MYB TF, Trifoliate (Tf), in young leaf primordia sustains morphogenetic activity, resulting in a surge in leaf dissection and inhibition of cell differentiation55. The tomato CLAU gene, another MYB TF, influences tomato leaf morphogenesis in a dose-dependent manner. Loss of function mutation (clau) causes extended morphogenesis and elaborate leaves, but over-expression of CLAU results in simplified leaves with primary leaflets55,97,98. Mutant of M. truncatula PHANTASTICA (MtPHAN), another MYB TF, is reported to exhibit leaf curling, deep serration of leaf margins and shortened petioles96. The association of IS-dependent repression of VunCCA1/VunLHY in the switch from compound leaves in the wild-type to unifoliate leaves in the UCL mutant is yet another evidence of the role of alternative splicing in regulating the functions of genes69,99,100. Although MYP TFs play diverse roles in plants101,102,103,104, our discovery of the likely implication of IS-dependent differential expression of VunCCA1/VunLHY in compound leaf development of a legume species is novel.
The insignificant expression of VunSTM, a KNOX gene, in the wild-type and its more than 4000-fold up-regulation in the UCL mutant, suggests that, under wild-type conditions, normal expressions of VunCCA1, VunLHY, VunLNK, VunRVE1, VunRVE4 and VunRVE7 either negatively regulate STM expression or replace its functions in cowpea primary morphogenesis, consistent with reports which have shown that KNOX genes do not express in the leaves of some legume species1,5.
Down-regulation of BRI1 results in BR-deficient-like phenotype in the UCL mutant
BRs are phytohormones which regulate various processes in plants, and CYP450 genes, notably CPD and CYP85A2, encode enzymes responsible for the biosynthesis of BRs42,46. CPD expression is predominantly regulated by the BRI1-dependent BR signaling pathway; bri1 mutant has an elevated expression of CPD, and the over-expression of BKI1, a BRI1 negative regulator, up-regulates CPD expression39,47,49. BRI1initiates the BR signaling cascade42, and BKI1 regulates plant architecture by negatively regulating BRI1 in the BR pathway49,105. Our differential gene expression analysis shows that BKI1 and CPD are up-regulated in the UCL mutant, while BRI1 and its positive regulator, BES1/BZR1, are down-regulated, producing the BR-deficient-like phenotypes, especially the curled dark green leaves and reduced plant size observed in the UCL mutant27,28. The differential expressions of these functionally related BR signaling pathway genes suggest two hypotheses. First, the identified BRI1 downstream variants in the UCL mutant compromise BRI1-dependent BR signaling pathway, resulting in the up-regulated expressions of CPD and BKI1. Second, repressed expressions of the components of the BRI1-dependent BR signaling is the output of the suppressed circadian clock endogenous oscillator of the UCL mutant76. The association of CPD with monooxygenase activity, iron ion binding, heme binding, and regulation of circadian rhythm46,106,107,108,109, some of the functionally enriched terms in the UCL mutant, strongly indicate that up-regulation of the VunCPD, and the corresponding down-regulation of BRI1 in the UCL mutant have roles in producing the mutant phenotype. Also, down-regulation of VunBRI1 is likely responsible for the functional depletion of response to red or far-red light and blue light signaling pathway in the UCL mutant49,51,110.
Taken together, our study has revealed important candidate genes, genomic variants and GO terms associated with the UCL mutant, laying a formidable foundation for future research that would aid in-depth understanding of the genetics of compound leaf development in cowpea. As our results suggest the involvement of various genes in the UCL mutant phenotype, future follow-up reverse genetic elucidation of the specific roles of the candidate genes, especially the candidates with genomic variants (VunLHY, VunRVE4, VunBRI1 and VunLOB), will provide useful insights into the molecular mechanisms underlying leaf morphogenesis in cowpea. Based on our results, we have stated three hypotheses as directions for future studies to build on our work. We strongly recommend the inclusion of de novo genome-wide IS analysis in forward genetic screens targeted at identifying genes or gene candidates responsible for spontaneous aberrant phenotypes in plants.
Methods
Plant materials and growth chamber conditions
We worked with three wild-type cowpea cultivars, IT86D-1010, IT97K-499–35 and Sasaque, reported in our speed breeding protocol111, and a spontaneous mutant segregant from a self-pollinated progeny of IT86D-1010, which we have named UCL mutant. As previously reported111, Sasaque, a Japanese cowpea cultivar, was originally obtained from TJ Higgins of Commonwealth Scientific and Industrial Research Organization, Australia; IT86D-1010 and IT97K-499–35 are cowpea breeding lines produced by the International Institute of Tropical Agriculture (IITA), Nigeria, and made available to us through the Hy-Gain for Smallholders Project (https://hy-gain.org/). Seeds of all the genotypes, including the UCL mutant, are maintained in the cowpea gene bank at ALRC, Tottori University, Japan. All the experimental plants were grown under the same controlled environmental conditions: 10 h photoperiod, 23 °C night/25 °C day temperature, and 70% day/75% night relative humidity.
Agro-morphological and genetic characterization of the UCL mutant
Agro-morphological variation between IT86D-1010, UCLHet and UCLHom was studied under the same growth conditions, with the genotypes laid out in a completely randomized design with three replications. Each replication consisted of three plants/genotype, and data were taken on total fresh shoot biomass, days to 50% flowering, pod length, number of pods/plant, number of seeds/pod and 100 seed weight. The mean and median of the data for each measured trait were either equal or nearly equal, indicating normality of the data. Using SPSS Statistics software (version 29.0), the data were subjected to analysis of variance, and significantly different means (p < 0.01) were separated using the Least Significant Difference Test. To determine whether the mutant locus is dominant or recessive, we observed segregation in both self- and cross-pollinated generations in our speed breeding facility. In the first self-pollinated generation, we observed 24 segregants harvested from the original mutant plant identified in the field. From the 24 segregants, we bulk-harvested seeds from UCLHet, UCLHom and revertant segregants. Using the UCLHet bulk, we cultivated and observed 200 plants in F2, and continued the same process until F6. With 50 seeds per generation, we also observed self-pollinated generations of UCLHom and revertant up to F6 for each of the genotypes. To observe allelic segregation in cross-pollinated populations, we made reciprocal crosses between the mutant genotypes (UCLHom and UCLHet) and the three wild-type cowpea cultivars mentioned under plant materials. This afforded us the opportunity of observing the transmission of the mutant locus in different genetic backgrounds.
RNA purification and sequencing
We extracted total RNA from frozen young leaves sampled from two weeks old seedlings of IT86D-1010 and confirmed self-reproducing UCLHom plants, using MagMAX Plant RNA Isolation Kit (Applied Biosystems). RNA quality was determined by NanoDrop 2000C spectrophotometer (Thermo Scientific), and quantification was done with Qubit 2.0 Fluorometer (Invitrogen). For each genotype, two bulked biological replicates, composed of RNA samples from 20 plants, were used for preparation of sequencing libraries. Strand-specific paired-end poly(A)-selected RNA-seq libraries of the bulked biological replicates prepared using TruSeq Stranded mRNA Library Prep Kit (Illumina) were sequenced with DNBSEQ-G400 (MGISEQ-2000RS) short reads sequencer.
RNA-seq data analysis
Before downstream applications, initial read quality assessment was done with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), after which the sequences were processed by fastp29. RNA STAR mapper30 was used to align the fastp-trimmed sequences to Vunguiculata_540_v1.0 reference genome, with Vunguiculata_540_v1.2 reference annotation as gene model (https://data.jgi.doe.gov/refine-download/phytozome?genome_id=540). We conducted a multi-sample 2-pass mapping, which enabled us to accurately discover and quantify splice junctions31. To analyze differential gene expression between the wild-type and UCL mutant, we applied featureCounts35 on the mapped collection of the datasets produced by RNA STAR mapper to obtain gene counts, after which DESeq236, was applied to conduct differential gene expression between the datasets. GO analysis of the DEGs was conducted with g:Profiler, a leading functional enrichment web resource37,38.
With the aid of StingTie63, we assembled and quantified the transcripts from the mapped collection of the libraries, guided by Vunguiculata_540_v1.0 reference genome and Vunguiculata_540_v1.2 reference annotation. The assembled transcripts were merged with Vunguiculata_540_v1.2 reference annotation to generate a novel non-redundant reference transcriptome annotation. Annotation accuracy was evaluated using GFFCompare64 to compare the merged reference annotation with Vunguiculata_540_v1.2 reference annotation. With the aid of IsoformSwitchAnalyzeR65, genome-wide AS was analyzed using the transcript-level gene expressions of all the transcripts in the datasets. We generated a raw switch list and their corresponding nucleotide and amino acid sequences, and used the amino acid and nucleotide sequences to, respectively, identify coding domains, and predict the coding potential of the of transcripts, using CPAT66. For all the RNA-seq data analyses reported here, we used RNA-seq data analysis tools either in the Europe (https://usegalaxy.eu/welcome/new) or USA (https://usegalaxy.org/) instance of Galaxy.
Prediction of expression and co-expression of candidate genes
Electronic gene browser at http://bar.utoronto.ca53,54 was applied to predict the developmental expression patterns of the gene candidates, and co-expression analysis was performed with ExpressionAngler at http://bar.utoronto.ca/ExpressionAngler/56.
Genome variant analysis
We studied the variants in the UCL mutant genome using shotgun sequences of purified bulked DNA samples of F2 segregants of IT97K-499–35 × UCLHom hybrid. NucleoMag Plant Genomic DNA Extraction Kit (Takara Bio USA) was used to extract and purify DNA samples from 10 seedlings each of UCLHet and UCLHom, and 20 seedlings of wild-type revertant. Combined DNA samples of UCLHet and UCLHom segregants constituted the UCL mutant bulk, and the DNA samples of the 20 wild-type revertant plants were bulked to create the revertant bulk. The two libraries, prepared using MGIEasy FS DNA Library Prep Set, were sequenced with DNBSEQ-G400 short reads sequencer. The sequence reads were processed with fastp29, after which the fastp-passed sequences were mapped to Vunguiculata_540_v1.0 reference genome, using Bowtie234. FreeBayes78 was applied to jointly call variant sites from the UCL mutant and revertant mapped genome sequences, and MiMohD VCF Filter79 was used to extract homozygous variant sites in the UCL mutant genome corresponding to homozygous wild-type loci in the revertant genome. The filtered variants were annotated using snpEff eff with a custom snpEff database built from Vunguiculata_540_v1.2 reference annotation, using snpEff build80. The variants were reported with MiMohD Report Variants79. All genomic data analyses reported here were conducted using genomic data analysis tools in the Europe instance of Galaxy (https://usegalaxy.eu/welcome/new).
Ethics declarations
As stated under the plant materials section, Sasaque was obtained from TJ Higgins of Commonwealth Scientific and Industrial Research Organization, Australia; IT86D-1010 and IT97K-499–35 were provided by IITA, Nigeria. The authors identified the original UCL mutant plant as a spontaneous mutant segregant from the self-pollinated progeny of IT86D-1010 in the 2021 cowpea field cultivation at ALRC. To confirm reproducibility and identity of the mutant, the seeds harvested from the original mutant plant were cultivated under controlled growth chamber conditions, and the self- and cross-pollinated populations were observed over generations. Seeds of all the genotypes reported here, including the UCL mutant, are maintained in the cowpea gene bank at ALRC, Tottori University, Japan, a subsidiary of the cowpea gene bank of Japan’s National Agriculture and Food Research Organization (https://www.gene.affrc.go.jp/index_en.php). The seeds and plants of the mutant are currently used for research purposes only, and handling and all the research activities are done in accordance with all relevant guidelines.
Data availability
All associated data and information, including short reads of RNA-seq and whole genome shotgun sequence data of the analyzed genotypes, not included in the main article are published as supplemental information linked with the main article.
References
Bharathan, G. et al. Homologies in leaf form inferred from KNOXI gene expression during development. Science 296, 1858–1860. https://doi.org/10.1126/science.1070343 (2002).
Efroni, I., Eshed, Y. & Lifschitz, E. Morphogenesis of simple and compound leaves: a critical review. Plant Cell 22, 1019–1032. https://doi.org/10.1105/tpc.109.073601 (2010).
He, L. et al. A molecular framework underlying the compound leaf pattern of Medicago truncatula. Nat. Plants 6, 511–521. https://doi.org/10.1038/s41477-020-0642-2 (2020).
Hake, S. et al. The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol 20, 125–151. https://doi.org/10.1146/annurev.cellbio.20.031803.093824 (2004).
Mo, X. et al. The genetic control of the compound leaf patterning in medicago truncatula. Front. Plant Sci. 12, 749989. https://doi.org/10.3389/fpls.2021.749989 (2021).
Nakayama, H., Leichty, A. R. & Sinha, N. R. Molecular mechanisms underlying leaf development, morphological diversification, and beyond. Plant Cell 34, 2534–2548. https://doi.org/10.1093/plcell/koac118 (2022).
Hay, A. & Tsiantis, M. The genetic basis for differences in leaf form between Arabidopsis thaliana and its wild relative Cardamine hirsuta. Nat. Genet. 38, 942–947. https://doi.org/10.1038/ng1835 (2006).
Du, L., Adkins, S. & Xu, M. Leaf development in medicago truncatula. Genes Basel https://doi.org/10.3390/genes13071203 (2022).
Hareven, D., Gutfinger, T., Parnis, A., Eshed, Y. & Lifschitz, E. The making of a compound leaf: Genetic manipulation of leaf architecture in tomato. Cell 84, 735–744. https://doi.org/10.1016/s0092-8674(00)81051-x (1996).
Bar, M. & Ori, N. Leaf development and morphogenesis. Development 141, 4219–4230. https://doi.org/10.1242/dev.106195 (2014).
Du, F., Guan, C. & Jiao, Y. molecular mechanisms of leaf morphogenesis. Mol. Plant 11, 1117–1134. https://doi.org/10.1016/j.molp.2018.06.006 (2018).
Rodriguez, R. E., Debernardi, J. M. & Palatnik, J. F. Morphogenesis of simple leaves: Regulation of leaf size and shape. Wiley Interdiscip. Rev. Dev. Biol 3, 41–57. https://doi.org/10.1002/wdev.115 (2014).
Chen, R. Functional genomics and genetic control of compound leaf development in medicago truncatula: An overview. Methods Mol. Biol. 197–203, 2018. https://doi.org/10.1007/978-1-4939-8633-0_14 (1822).
Wang, H. et al. Control of compound leaf development by FLORICAULA/LEAFY ortholog SINGLE LEAFLET1 in Medicago truncatula. Plant Physiol. 146, 1759–1772. https://doi.org/10.1104/pp.108.117044 (2008).
Jiao, K. et al. Genetic control of compound leaf development in the mungbean (Vigna radiata L). Hortic. Res. 6, 23. https://doi.org/10.1038/s41438-018-0088-0 (2019).
Wojciechowski, M. F., Sanderson, M.J., Steele, K.P., & Liston, A. in Advances in Legume Systematics Vol. 9 (ed P.S. Herendeen, Bruneau, A., and Pollard, P. S. ) 277 - 298 ( Royal Botanic Gardens, Kew, 2000).
Duan, L. et al. Phylogenomic framework of the IRLC legumes (Leguminosae subfamily Papilionoideae) and intercontinental biogeography of tribe Wisterieae. Mol. Phylogenet. Evol. 163, 107235. https://doi.org/10.1016/j.ympev.2021.107235 (2021).
Champagne, C. E. et al. Compound leaf development and evolution in the legumes. Plant Cell 19, 3369–3378. https://doi.org/10.1105/tpc.107.052886 (2007).
Xia, Z. et al. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 109, E2155-2164. https://doi.org/10.1073/pnas.1117982109 (2012).
Lu, S. et al. Stepwise selection on homeologous PRR genes controlling flowering and maturity during soybean domestication. Nat. Genet. 52, 428–436. https://doi.org/10.1038/s41588-020-0604-7 (2020).
Han, J. et al. Creation of early flowering germplasm of soybean by CRISPR/Cas9 technology. Front. Plant. Sci. 10, 1446. https://doi.org/10.3389/fpls.2019.01446 (2019).
Li, Y. et al. The legume-specific transcription factor E1 controls leaf morphology in soybean. BMC Plant. Biol. 21, 531. https://doi.org/10.1186/s12870-021-03301-1 (2021).
Singh, B. B. & IITA. Cowpea: The Food Legume of the 21st Century. (Crop Science Society of America, 2014).
Lonardi, S. et al. The genome of cowpea (Vigna unguiculata [L.] Walp). Plant J. 98, 767–782. https://doi.org/10.1111/tpj.14349 (2019).
Rawal, K. M., Porter, W. M., Franckowiak, J. D., Fawole, I. & Rachie, K. O. Unifoliolate leaf: A mutant in cowpeas. Heridity 67, 193–194. https://doi.org/10.1093/oxfordjournals.jhered.a108707 (1976).
Fawole, I. Genetic analysis of mutations at loci controlling leaf form in cowpea (Vigna unguiculata. J. Hered. 92, 43–50. https://doi.org/10.1093/jhered/92.1.43 (2001).
Li, J. M., Nagpal, P., Vitart, V., McMorris, T. C. & Chory, J. A role for brassinosteroids in light-dependent development of Arabidopsis. Science 272, 398–401. https://doi.org/10.1126/science.272.5260.398 (1996).
Ohnishi, T. et al. CYP90A1/CPD, a brassinosteroid biosynthetic cytochrome P450 of Arabidopsis, catalyzes C-3 oxidation. J. Biol. Chem. 287, 31551–31560. https://doi.org/10.1074/jbc.M112.392720 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. https://doi.org/10.1093/bioinformatics/bts635 (2013).
Veeneman, B. A., Shukla, S., Dhanasekaran, S. M., Chinnaiyan, A. M. & Nesvizhskii, A. I. Two-pass alignment improves novel splice junction quantification. Bioinformatics 32, 43–49. https://doi.org/10.1093/bioinformatics/btv642 (2016).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. https://doi.org/10.1093/bioinformatics/btw354 (2016).
Wang, L., Wang, S. & Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. https://doi.org/10.1093/bioinformatics/bts356 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. https://doi.org/10.1093/bioinformatics/btt656 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Kolberg, L. et al. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 51, W207–W212. https://doi.org/10.1093/nar/gkad347 (2023).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193-200. https://doi.org/10.1093/nar/gkm226 (2007).
Goda, H., Shimada, Y., Fujioka, S. & Yoshida, S. Classification of brassinosteroid-regulated genes based on expression profiles in bri1 and in response to a protein kinase inhibitor, staurosporin. Biosci. Biotechnol. Biochem. 68, 1605–1607. https://doi.org/10.1271/bbb.68.1605 (2004).
Chen, J. et al. Conserved genetic determinant of motor organ identity in Medicago truncatula and related legumes. Proc. Natl. Acad. Sci. USA 109, 11723–11728. https://doi.org/10.1073/pnas.1204566109 (2012).
Zhou, C. et al. Identification and characterization of petiolule-like pulvinus mutants with abolished nyctinastic leaf movement in the model legume Medicago truncatula. New Phytol. 196, 92–100. https://doi.org/10.1111/j.1469-8137.2012.04268.x (2012).
Kong, Y. et al. Brassinosteroid homeostasis is critical for the functionality of the Medicago truncatula pulvinus. Plant. Physiol. 185, 1745–1763. https://doi.org/10.1093/plphys/kiab008 (2021).
Bell, E. M. et al. LATERAL ORGAN BOUNDARIES negatively regulates brassinosteroid accumulation to limit growth in organ boundaries. Proc. Natl. Acad. Sci. USA 109, 21146–21151. https://doi.org/10.1073/pnas.1210789109 (2012).
Clouse, S. D. Brassinosteroids. Arabidopsis Book 1, e0009. https://doi.org/10.1199/tab.0009 (2002).
Shimada, Y. et al. Brassinosteroid-6-oxidases from Arabidopsis and tomato catalyze multiple C-6 oxidations in brassinosteroid biosynthesis. Plant Physiol. 126, 770–779. https://doi.org/10.1104/pp.126.2.770 (2001).
Bancos, S. et al. Diurnal regulation of the brassinosteroid-biosynthetic CPD gene in Arabidopsis. Plant Physiol. 141, 299–309. https://doi.org/10.1104/pp.106.079145 (2006).
Bancos, S. et al. Regulation of transcript levels of the Arabidopsis cytochrome P450 genes involved in brassinosteroid biosynthesis. Plant Physiol. 130, 504–513. https://doi.org/10.1104/pp.005439 (2002).
Mathur, J. et al. Transcription of the Arabidopsis CPD gene, encoding a steroidogenic cytochrome P450, is negatively controlled by brassinosteroids. Plant. J. 14, 593–602. https://doi.org/10.1046/j.1365-313x.1998.00158.x (1998).
Wang, X. L. & Chory, J. Brassinosteroids regulate dissociation of BKI1, a negative regulator of BRI1 signaling, from the plasma membrane. Science 313, 1118–1122. https://doi.org/10.1126/science.1127593 (2006).
Jaillais, Y. & Vert, G. Brassinosteroid signaling and BRI1 dynamics went underground. Curr. Opin. Plant Biol. 33, 92–100. https://doi.org/10.1016/j.pbi.2016.06.014 (2016).
Clouse, S. D. Brassinosteroid signal transduction: From receptor kinase activation to transcriptional networks regulating plant development. Plant Cell 23, 1219–1230. https://doi.org/10.1105/tpc.111.084475 (2011).
Jiang, J. J., Zhang, C. & Wang, X. L. A recently evolved isoform of the transcription factor BES1 promotes brassinosteroid signaling and development in. Plant Cell 27, 361–374. https://doi.org/10.1105/tpc.114.133678 (2015).
Libault, M. et al. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant. J. 63, 86–99. https://doi.org/10.1111/j.1365-313X.2010.04222.x (2010).
Winter, D. et al. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS One 2, e718. https://doi.org/10.1371/journal.pone.0000718 (2007).
Naz, A. A. et al. Trifoliate encodes an MYB transcription factor that modulates leaf and shoot architecture in tomato. Proc. Natl. Acad. Sci. USA 110, 2401–2406. https://doi.org/10.1073/pnas.1214300110 (2013).
Austin, R. S. et al. New BAR tools for mining expression data and exploring Cis-elements in Arabidopsis thaliana. Plant. J. 88, 490–504. https://doi.org/10.1111/tpj.13261 (2016).
Kim, H., Kim, Y., Yeom, M., Lim, J. & Nam, H. G. Age-associated circadian period changes in Arabidopsis leaves. J. Exp. Bot. 67, 2665–2673. https://doi.org/10.1093/jxb/erw097 (2016).
Rawat, R. et al. REVEILLE8 and PSEUDO-REPONSE REGULATOR5 form a negative feedback loop within the Arabidopsis circadian clock. PLoS Genet. 7, e1001350. https://doi.org/10.1371/journal.pgen.1001350 (2011).
Sorkin, M. L. et al. COLD REGULATED GENE 27 and 28 antagonize the transcriptional activity of the RVE8/LNK1/LNK2 circadian complex. Plant. Physiol. 192, 2436–2456. https://doi.org/10.1093/plphys/kiad210 (2023).
Zhang, X. B. et al. Constitutive expression of CIR1 (RVE2) affects several circadian-regulated processes and seed germination in Arabidopsis. Plant. J. 51, 512–525. https://doi.org/10.1111/j.1365-313X.2007.03156.x (2007).
Rawat, R. et al. REVEILLE1, a Myb-like transcription factor, integrates the circadian clock and auxin pathways. Proc. Natl. Acad. Sci. USA 106, 16883–16888. https://doi.org/10.1073/pnas.0813035106 (2009).
Liu, Z. et al. Characterization of the REVEILLE family in Rosaceae and role of PbLHY in flowering time regulation. BMC Genom. 24, 49. https://doi.org/10.1186/s12864-023-09144-4 (2023).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. https://doi.org/10.1038/nbt.3122 (2015).
Pertea, G., Pertea, M. GFF utilities: GffRead and GffCompare. Res https://doi.org/10.12688/f1000research.23297.2 (2020)
Vitting-Seerup, K. & Sandelin, A. IsoformSwitchAnalyzeR: Analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics 35, 4469–4471. https://doi.org/10.1093/bioinformatics/btz247 (2019).
Wang, L. et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74. https://doi.org/10.1093/nar/gkt006 (2013).
Rayson, S. et al. A role for nonsense-mediated mRNA decay in plants: Pathogen responses are induced in Arabidopsis thaliana NMD mutants. PLoS One 7, e31917. https://doi.org/10.1371/journal.pone.0031917 (2012).
Udy, D.B., Bradley, R.K. Nonsense-mediated mRNA decay uses complementary mechanisms to suppress mRNA and protein accumulation. Life Sci Alliance https://doi.org/10.26508/lsa.202101217 (2022)
Syed, N. H., Kalyna, M., Marquez, Y., Barta, A. & Brown, J. W. Alternative splicing in plants–coming of age. Trends Plant. Sci. 17, 616–623. https://doi.org/10.1016/j.tplants.2012.06.001 (2012).
Nogueira, G., Fernandes, R., Garcia-Moreno, J. F. & Romao, L. Nonsense-mediated RNA decay and its bipolar function in cancer. Mol. Cancer 20, 72. https://doi.org/10.1186/s12943-021-01364-0 (2021).
Lindeboom, R. G. H., Vermeulen, M., Lehner, B. & Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. https://doi.org/10.1038/s41588-019-0517-5 (2019).
Lindeboom, R. G., Supek, F. & Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 48, 1112–1118. https://doi.org/10.1038/ng.3664 (2016).
Caux-Moncoutier, V. et al. Impact of BRCA1 and BRCA2 variants on splicing: Clues from an allelic imbalance study. Eur. J. Hum. Genet. 17, 1471–1480. https://doi.org/10.1038/ejhg.2009.89 (2009).
Park, M. J., Kwon, Y. J., Gil, K. E. & Park, C. M. LATE ELONGATED HYPOCOTYL regulates photoperiodic flowering via the circadian clock in Arabidopsis. BMC Plant. Biol. 16, 114. https://doi.org/10.1186/s12870-016-0810-8 (2016).
Mizoguchi, T. et al. LHY and CCA1 are partially redundant genes required to maintain circadian rhythms in Arabidopsis. Dev. Cell 2, 629–641. https://doi.org/10.1016/s1534-5807(02)00170-3 (2002).
Venkat, A., Muneer, S. Role of circadian rhythms in major plant metabolic and signaling pathways. Front. Plant Sci 13, 836244. https://doi.org/10.3389/fpls.2022.836244 (2022)
Harmer, S. L. The circadian system in higher plants. Annu. Rev. Plant. Biol. 60, 357–377. https://doi.org/10.1146/annurev.arplant.043008.092054 (2009).
Garrison, E. A. M., G. Haplotype-based variant detection from short-read sequencing, https://arxiv.org/abs/1207.3907v2 (2012).
Maier, W., Moos, K., Seifert, M., & Baumeister, R. MiModD - Mutation Identification in Model Organism Genomes, https://sourceforge.net/projects/mimodd/ (2014).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly 6, 80–92. https://doi.org/10.4161/fly.19695 (2012).
Bendix, C., Marshall, C. M. & Harmon, F. G. Circadian clock genes universally control key agricultural traits. Mol. Plant. 8, 1135–1152. https://doi.org/10.1016/j.molp.2015.03.003 (2015).
Inoue, K., Araki, T. & Endo, M. Integration of input signals into the gene network in the plant circadian clock. Plant. Cell Physiol. 58, 977–982. https://doi.org/10.1093/pcp/pcx066 (2017).
Creux, N. & Harmer, S. Circadian rhythms in plants. Cold Spring Harb Perspect. Biol. https://doi.org/10.1101/cshperspect.a034611 (2019).
McClung, C. R. Circadian rhythms in plants. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 52, 139–162. https://doi.org/10.1146/annurev.arplant.52.1.139 (2001).
Schaffer, R. et al. The late elongated hypocotyl mutation of Arabidopsis disrupts circadian rhythms and the photoperiodic control of flowering. Cell 93, 1219–1229. https://doi.org/10.1016/s0092-8674(00)81465-8 (1998).
Wang, Z. Y. & Tobin, E. M. Constitutive expression of the CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) gene disrupts circadian rhythms and suppresses its own expression. Cell 93, 1207–1217. https://doi.org/10.1016/s0092-8674(00)81464-6 (1998).
Strayer, C. et al. Cloning of the Arabidopsis clock gene TOC1, an autoregulatory response regulator homolog. Science 289, 768–771. https://doi.org/10.1126/science.289.5480.768 (2000).
Makino, S., Matsushika, A., Kojima, M., Oda, Y. & Mizuno, T. Light response of the circadian waves of the APRR1/TOC1 quintet: when does the quintet start singing rhythmically in Arabidopsis?. Plant Cell Physiol. 42, 334–339. https://doi.org/10.1093/pcp/pce036 (2001).
Fujiwara, S. et al. Circadian clock proteins LHY and CCA1 regulate SVP protein accumulation to control flowering in Arabidopsis. Plant. Cell 20, 2960–2971. https://doi.org/10.1105/tpc.108.061531 (2008).
Carre, I. A. & Kim, J. Y. MYB transcription factors in the Arabidopsis circadian clock. J. Exp. Bot. 53, 1551–1557. https://doi.org/10.1093/jxb/erf027 (2002).
Kugan, H. M. et al. Circadian rhythms in legumes: What do we know and what else should we explore?. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22094588 (2021).
Weiss, J. et al. Diel pattern of circadian clock and storage protein gene expression in leaves and during seed filling in cowpea (Vigna unguiculata). BMC Plant. Biol. 18, 33. https://doi.org/10.1186/s12870-018-1244-2 (2018).
Bar, M. et al. CLAUSA is a MYB transcription factor that promotes leaf differentiation by attenuating cytokinin signaling. Plant Cell 28, 1602–1615. https://doi.org/10.1105/tpc.16.00211 (2016).
Schmitz, G. et al. The tomato Blind gene encodes a MYB transcription factor that controls the formation of lateral meristems. Proc. Natl. Acad. Sci. USA 99, 1064–1069. https://doi.org/10.1073/pnas.022516199 (2002).
Busch, B. L. et al. Shoot branching and leaf dissection in tomato are regulated by homologous gene modules. Plant Cell 23, 3595–3609. https://doi.org/10.1105/tpc.111.087981 (2011).
Ge, L., Peng, J., Berbel, A., Madueno, F. & Chen, R. Regulation of compound leaf development by PHANTASTICA in Medicago truncatula. Plant Physiol. 164, 216–228. https://doi.org/10.1104/pp.113.229914 (2014).
Moore, I., Galweiler, L., Grosskopf, D., Schell, J. & Palme, K. A transcription activation system for regulated gene expression in transgenic plants. Proc. Natl. Acad. Sci. U S A 95, 376–381. https://doi.org/10.1073/pnas.95.1.376 (1998).
Shani, E. et al. Stage-specific regulation of Solanum lycopersicum leaf maturation by class 1 KNOTTED1-LIKE HOMEOBOX proteins. Plant Cell 21, 3078–3092. https://doi.org/10.1105/tpc.109.068148 (2009).
Tu, M. & Li, Y. Profiling alternative 3’ untranslated regions in sorghum using RNA-seq data. Front. Genet. 11, 556749. https://doi.org/10.3389/fgene.2020.556749 (2020).
Srivastava, A. K., Lu, Y., Zinta, G., Lang, Z. & Zhu, J. K. UTR-dependent control of gene expression in plants. Trends Plant. Sci. 23, 248–259. https://doi.org/10.1016/j.tplants.2017.11.003 (2018).
Baldoni, E., Genga, A. & Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 16, 15811–15851. https://doi.org/10.3390/ijms160715811 (2015).
Komatsuzaki, A., Hoshino, A., Otagaki, S., Matsumoto, S. & Shiratake, K. Genome-wide analysis of R2R3-MYB transcription factors in Japanese morning glory. PLoS One 17, e0271012. https://doi.org/10.1371/journal.pone.0271012 (2022).
Li, P. et al. Diverse roles of MYB transcription factors in regulating secondary metabolite biosynthesis, shoot development, and stress responses in tea plants (Camellia sinensis). Plant J. 110, 1144–1165. https://doi.org/10.1111/tpj.15729 (2022).
Wang, X., Niu, Y., Zheng, Y. Multiple functions of MYB transcription factors in abiotic stress responses. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22116125 (2021)
Wang, D. X. et al. BKI1 regulates plant architecture through coordinated inhibition of the brassinosteroid and ERECTA signaling pathways in. Mol. Plant 10, 297–308. https://doi.org/10.1016/j.molp.2016.11.014 (2017).
Degtyarenko, K. N. Structural domains of P450-containing monooxygenase systems. Protein Eng. 8, 737–747. https://doi.org/10.1093/protein/8.8.737 (1995).
McLean, K. J., Girvan, H. M. & Munro, A. W. Cytochrome P450/redox partner fusion enzymes: Biotechnological and toxicological prospects. Exp. Opin. Drug Met. 3, 847–863. https://doi.org/10.1517/17425255.3.6.847 (2007).
Munro, A. W., Girvan, H. M. & McLean, K. J. Cytochrome P450 - redox partner fusion enzymes. Bba-Gen Subjects 1770, 345–359. https://doi.org/10.1016/j.bbagen.2006.08.018 (2007).
Zhan, H. et al. OsCPD1 and OsCPD2 are functional brassinosteroid biosynthesis genes in rice. Plant. Sci. 325, 111482. https://doi.org/10.1016/j.plantsci.2022.111482 (2022).
Wang, Z. Y., Bai, M. Y., Oh, E. & Zhu, J. Y. Brassinosteroid signaling network and regulation of photomorphogenesis. Annu. Rev. Genet. 46, 701–724. https://doi.org/10.1146/annurev-genet-102209-163450 (2012).
Edet, O. U. & Ishii, T. Cowpea speed breeding using regulated growth chamber conditions and seeds of oven-dried immature pods potentially accommodates eight generations per year. Plant Methods 18, 106. https://doi.org/10.1186/s13007-022-00938-3 (2022).
Author information
Authors and Affiliations
Contributions
All the authors designed the study, made the required crosses, and evaluated the progenies. T. I. and B. E. U. analyzed and reported the agronomic data. O. U. E. analyzed and interpreted the RNA-seq and genome sequence datasets and wrote the manuscript. All the authors reviewed and approved the manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Edet, O.U., Ubi, B.E. & Ishii, T. Genomic analysis of a spontaneous unifoliate mutant reveals gene candidates associated with compound leaf development in Vigna unguiculata [L] Walp. Sci Rep 14, 10654 (2024). https://doi.org/10.1038/s41598-024-61062-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-61062-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
