
Abstract
Dopamine-dependent long-term plasticity is believed to be a cellular mechanism underlying reinforcement learning. In response to reward and reward-predicting cues, phasic dopamine activity potentiates the efficacy of corticostriatal synapses on spiny projection neurons (SPNs). Since phasic dopamine activity also encodes other behavioural variables, it is unclear how postsynaptic neurons identify which dopamine event is to induce long-term plasticity. Additionally, it is unknown how phasic dopamine released from arborised axons can potentiate targeted striatal synapses through volume transmission. To examine these questions we manipulated striatal cholinergic interneurons (ChIs) and dopamine neurons independently in two distinct in vivo paradigms. We report that long-term potentiation (LTP) at corticostriatal synapses with SPNs is dependent on the coincidence of pauses in ChIs and phasic dopamine activation, critically accompanied by SPN depolarisation. Thus, the ChI pause defines the time window for phasic dopamine to induce plasticity, while depolarisation of SPNs constrains the synapses eligible for plasticity.
Similar content being viewed by others


Introduction
In the wild, animals need to take particular actions to maximise reward in a given situation. After obtaining a reward, or a sensory cue that predicts a reward, midbrain dopamine neurons briefly increase their firing rate in synchrony1. In reinforcement learning, phasic dopamine activity is thought to reinforce the actions that led to a reward to increase the chances of earning the same reward in future2,3. The cellular mechanism of reinforcement learning is believed to involve dopamine-dependent long-term plasticity, where increased dopamine levels enhance the efficacy of glutamatergic synapses between cortex and striatum4,5,6. Thus, striatal spiny projection neurons (SPNs) involved in executing a rewarded action will respond more effectively to subsequent presentations of the same cortical input pattern, in order to maximise future reward delivery. However, dopamine neurons not only increase their firing rate after reward and reward-predicting cues, but also increase to other encoded variables such as distance to reward7,8, movement9,10, and behavioural choices11, although these variables may be encoded in a smaller discrete population of dopamine neurons12. Therefore, it is unclear how corticostriatal synapses might identify the appropriate dopamine signal from which to induce long-term plasticity during reinforcement learning.
In the current study, we tested the previously-proposed hypothesis13,14,15,16,17,18 that the striatal tonically active neurons (TANs), likely to be ChIs, play a critical role in determining which dopamine signal is involved in modulating synaptic transmission. Although only 1% of striatal neurons are ChIs, they can regulate SPNs directly19 and indirectly20,21, and can modulate axonal dopamine release during coincident phasic dopamine activity22,23,24,25. ChIs exhibit excitation-pause-rebound multiphasic activity in response to reward and reward-indicating cues26,27,28. Critically, the pause phase coincides with phasic dopamine activity during learning14,15. Therefore, the ChI pause has been suggested to facilitate dopamine-dependent plasticity13,14,15,16,17,18. However, this hypothesis has never been tested in an intact brain in vivo due to the challenge of inducing synchronised pauses in sparsely distributed ChIs, aligned with phasic activity of dopamine neurons, within a physiologically meaningful timescale.
We recently showed that the firing of ChIs is entrained to fluctuations in excitatory input29. This observation enables us to test for the first time whether the ChI multiphasic response is involved in dopamine-dependent plasticity. Using the inverted striatal local field potential (iLFP) as a read-out of spontaneous and electrically evoked excitatory activity in our anaesthetised preparation29, we are able to manipulate ChIs with cortical stimulation at the same time as we manipulate dopamine neurons. Here, we hypothesised that a temporal coincidence of a pause in the multiphasic activity of ChIs and phasic dopamine is required to induce long-term potentiation of corticostriatal synapses. Further, we propose that a marginal shift of the timing of phasic dopamine activity (a few hundred milliseconds) to overlap with excitation phases of ChI will switch the direction of long-term plasticity.
Another currently unresolved question in dopamine-dependent plasticity is how widespread changes in dopamine signalling in the striatum induce long-term plasticity only at those synapses involved in driving actions that lead to reward. Dopamine neurons have extensively arborised axons such that a single neuron can cover up to 5% of the striatum30. Furthermore, the volume transmission of dopamine in the striatum will further enhance the opportunity for synchronised phasic dopamine activity to elevate dopamine levels at synapses irrelevant to the rewarded action. One possible mechanism for constraining synaptic change is to limit plasticity to those postsynaptic neurons depolarised during the action. In slice preparations, depolarisation of the postsynaptic neurons is necessary to form long-term plasticity31 and a conjunction of pre- and postsynaptic activity, plus a reward signal (a three-factor plasticity rule) has been proposed5. However, the role of depolarisation has not been adequately characterised in vivo. Here, we further hypothesise that depolarisation of the postsynaptic SPNs is necessary for the induction of long-term plasticity, and the function is to constrain dopamine-dependent plasticity to the targeted synapses.
To test these hypotheses, we undertook two distinct in vivo experiments where we manipulated both midbrain dopamine neurons and putative ChIs (pChIs) so that phasic dopamine was present at different phases of the ChI multiphasic response. In addition, in the second set of experiments we also manipulated the membrane potential of the postsynaptic SPN by injecting positive current intracellularly, to determine the optimal timing of depolarisation for long-term plasticity. Our results suggest that the optimal conditions for induction of in vivo long-term potentiation at corticostriatal synapses on SPNs are a coincidence of phasic activity in dopamine neurons, pauses in ChIs, and depolarisation of postsynaptic SPNs.
Results
Two experimental paradigms were used to investigate the integration and timing of glutamatergic, dopaminergic, and cholinergic signals in the striatum.
The coincidence of ChI pause and phasic dopamine is required to induce LTP
In the first experimental paradigm, we made single-unit extracellular recordings of SPNs in urethane-anaesthetised rats (Fig. 1a, b, Supplementary Fig. 1). Contralateral cortical electrical test pulses were applied to elicit spikes in SPNs, and a change in the probability of cortically-evoked spikes was used to quantify corticostriatal plasticity. In addition to evoking spikes in SPNs, the cortical stimulation also entrained the firing of striatal ChIs (Fig. 1c), with the pause indicated by the ‘receding phase’ and maximal firing during the ‘rising phase’ of the iLFP, as described previously29,32 (Fig. 1c).

a Cortical stimulation was applied to elicit spike activity in SPNs and to entrain the activity of pChIs. Contralateral visual stimulation was applied with BIC injected into the SC to activate dopamine neurons and depolarise SPNs. b A representative example from 11 labelled SPNs with spine arrowed in inset (Scale bar 20 µm; inset 5 µm). c Upper, Cortical stimulation alone entrained the firing pattern of a pChI (orange raster and histogram). The excitation phase of the pChI occurs at the rising phase of the iLFP (orange trace), and the pause phase occurs at receding phases of the iLFP. Superimposed is the pChI response to the visual stimulation alone (green) with the SC disinhibited, and the likely period of released dopamine following visual activation indicated (purple). The visual stimulation is aligned to the timing used for pairing in the lower panels to illustrate the effect of the alignment. Because the excitation induced by the visual stimulation is very slow, it did not interfere with the pause induced by the critical stimulation. Lower, pairing cortical stimulation and visual stimulation at different intervals can drive phasic dopamine activity to coincide with either a ChI pause (left) or excitation phase (right). Note, due to the difficulties of recording pChI in vivo, three different pChIs were recorded to demonstrate how cortical stimulation alone (upper, orange), or visual stimulation alone (upper, green), or pairing of two stimuli (lower) regulate the firing pattern of pChIs, respectively.
Dopamine neurons were activated phasically by a physiologically relevant stimulation, a light flash to the contralateral eye when the superior colliculus (SC) was disinhibited by local injection of the GABA antagonist bicuculline (BIC; Fig. 1a). During collicular disinhibition, a visual stimulus drives phasic activity in dopamine neurons at about 110 ms after a light flash33,34 (Supplementary Fig. 2), as well as contributing to a slower response (latency ~200 ms) in pChIs (Fig. 1c). Thus, by pairing cortical stimulation with disinhibited visual stimulation at varying intervals, we were able to activate phasic dopamine activity at the timing of either a pause or excitation of pChIs, respectively (Fig. 1c).
To determine whether the coincidence of the ChI pause and phasic dopamine is required for potentiation in SPNs, we first acquired baseline cortical responses by applying cortical test stimulation alone for 10 min at 0.2 Hz (see Fig. 2b for paradigm). Then, to induce synaptic plasticity, cortical stimulation was paired with light stimulation. After 5 min of pairing, the SC was disinhibited locally by BIC and then pairing continued for another 10 min. The combination of these two manipulations either drove phasic dopamine activity to coincide with a pChI pause (the ‘Match’ group) or with pChI excitation (‘Mismatch’ group). Then, spike activity of SPNs in response to cortical test stimulation alone was continued for at least another 30 mins. We found that the spike activity of SPNs induced by cortical stimulation (total number of spikes at short-latency; < 30 ms) remained potentiated only when the period of pChI pauses and phasic dopamine was coincident (‘Match’ group, Fig. 2a left and 2b). In contrast, depression of spike activity occurred in the ‘Mismatch’ group (Fig. 2a right and 2b), in which dopamine was temporally separated from the pChI pause.

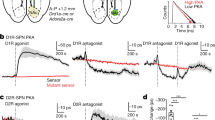
a Cortical stimulation induced spike activity in SPNs. A representative potentiation (left) and depression (right) in SPNs 30 min after match or mismatch pairings, respectively. b Spike activity in SPNs (mean ± S.E.M.) was potentiated when the light-induced dopamine signal was coincident with ChI pauses (red, p = 0.0227), but not when coincident with excitation of ChIs (light blue, p = 0.0349) or when phasic dopamine was absent (dark blue—no light flashes after BIC, p = 0. 2219). N = 6 neurons in each group, *p < 0.05 (two-tailed paired t test; averaged last 10 min compared to baseline).
We further tested whether the pChI pause alone can induce potentiation. In the control group (‘Control’), light flashes were only paired with cortical stimulation for the 5 min prior to bicuculline injection and not subsequently, so pChI pauses were entrained by the cortical stimulation but dopamine neurons were not phasically-activated. We found a similar depression in the ‘Control’ group as the ‘Mismatch’ group (Fig. 2b). Thus, pChI pauses elicited by cortical stimulation were insufficient to induce potentiation without phasic dopamine. Therefore, potentiation of corticostriatal responses only emerged when phasic dopamine was coincident with the pChI pause.
However, using this particular experimental paradigm we were unable to test our second hypothesis regarding the necessity of postsynaptic depolarisation for corticostriatal plasticity in vivo. In this preparation, SPNs were depolarised by the light stimulation via the tecto-thalamo-striatal glutamatergic pathway at a similar time as dopamine neurons were activated (Fig. 1a)35,36. Therefore, we used a second experimental paradigm to allow us to manipulate SPN depolarisation independently of SNc dopamine cell activation (Fig. 3a, b).

a Cortical stimulation induced PSPs in SPNs. Dopamine neurons were activated by electrical stimulation and depolarisation was induced by intracellular current injection. b A representative example from 11 labelled SPNs (Scale bar 20 µm; inset 2 µm). c Electrical stimulation of the SNc was set to match (purple shade) or mismatch (blue shade) the ChI pause, determined from the striatal iLFP. d Example corticostriatal PSPs potentiated (red) or depressed (light blue) in slope (expanded in insets) and in amplitude 20 min after baseline (black traces) when SNc stimulation matched or mismatched the ChI pause, respectively. e Group average effect on PSPs of SNc stimulation matched (N = 7) vs mismatched (N = 7); **p < 0.01 (Mean ± S.E.M.; unpaired t test at 20 min, p = 0.0025). f One-way ANOVA with Brown-Forsythe and Welch ANOVA post hoc tests to compare Match group (which induced LTP on average) to all other groups (where no change or LTD was induced): Match vs. Mismatch p = 0.008, Match vs. Dissociated (N = 6) p = 0.008, Match vs. SNc stim only (N = 5) p = 0.0005, Match vs. Depol-only (N = 7) p = 0.0010).
The depolarisation of postsynaptic SPNs is also required for potentiation
We tested whether depolarisation of postsynaptic SPNs is required for LTP in vivo by using a paradigm with which we have previously demonstrated an association between synaptic plasticity and positive reinforcement4. In these experiments, we applied a contralateral electrical test stimulation to simulate cortical input, and directly activated dopamine neurons with electrical stimulation in the midbrain. Intracellular recordings were made to depolarise the postsynaptic SPNs and measure the postsynaptic potential (PSP).
In our previous study4 we found that dopamine-dependent long-term potentiation (LTP) of synapses on recorded SPNs was induced when the neurons were depolarised during the SPN ‘down state’ using intracellular current injection, and when dopamine neurons were activated simultaneously. However, according to our recent investigation of the factors underlying the firing activity of ChIs29, the down state of SPNs not only corresponds to a period of minimum excitatory cortical input to the striatum37, but also to the time when ChIs exhibit a pause (Fig. 3c). Therefore, serendipitously, the LTP in our previous study was elicited by activation of dopamine neurons and the depolarisation of SPNs during the period of a pChI pause. However, the necessity of each of the factors contributing to LTP were not further elucidated in that study.
Here we used a similar paradigm to determine the temporal requirements for activation of dopamine neurons, the pChI pause and depolarisation of SPNs for the induction of corticostriatal LTP. We first successfully replicated our previous finding of LTP induction by applying the same combination of dopamine neuron stimulation and depolarisation in the down state, which we now know corresponds to the ChI pause, in naïve animals (Match: 17.2 ± 10.2% at +20 min, N = 7 animals; Fig. 3c–e).
We then tested if the presence of the ChI pause was necessary for the induction of LTP. In contrast, we found that when SPN depolarisation and dopamine input were applied at the time that ChIs are most excited, during the spontaneous SPN up state, that no significant change in synaptic efficacy resulted (Mismatch: −1.0 ± 5.1% at 20 min, N = 7 animals; Fig. 3e). Notably, the difference in plasticity resulting in the Match and the Mismatch group was not due to differences in the level of membrane depolarisation achieved, since current injection in both groups was set to just exceed the threshold for action potential firing (see Methods). Thus, these results agree with the extracellular experiments in demonstrating a need for dopamine activation to coincide with the pause in ChIs to induce lasting potentiation.
We further tested whether the combination of dopamine and the pause in ChIs, or the combination of SPN depolarisation and the pause in pChIs, is effective in inducing LTP. We found that either dopamine stimulation alone (SNc stim only: −14.0 ± 4.4% at +20 min, N = 5 animals; p < 0.001 in comparison to match group) or depolarisation of SPNs alone (Depol only: −15.8 ± 6.1% at 20 min, N = 7 animals; p < 0.01 in comparison to the match group) applied during the pChI pause period, induced LTD.
Finally, we tested whether coincident SPN depolarisation and dopamine signalling are required for LTP induction. We temporally separated the depolarisation and dopamine stimulation. We applied the depolarisation immediately following SPN up state initiation, at the excitation phase of pChIs, and dopamine input was applied in the following down state corresponding to the pause phase in ChIs. We found that temporally dissociating these components led to no change in synaptic efficacy (Dissociated: −2.7 ± 6.6% at 20 min, N = 6 animals; p < 0.01 in comparison to match group, Fig. 3c, f).
Discussion
Despite differences in recording techniques and measures of plasticity, results from both extracellular and intracellular recordings were in agreement, that temporal coincidence of phasic dopamine, pChI pause and SPN depolarisation is required for the induction of corticostriatal LTP in vivo. No change or synaptic depression is found if these signals occur during the excitation phase of the pChIs, if dopamine signalling or SPN depolarisation occur alone during the pause phase, or if they are separated in time around the pChI pause (Fig. 4).

Corticostriatal LTP (pink) is induced when SPNs are depolarised (brown) in a striatal area where a ChI pause response (green) is coincident with a phasic dopamine signal (blue). Other conditions will result in LTD or no change (NC).
Our results suggest that the ChI pause may define a time window for phasic dopamine to induce long-term synaptic potentiation. We here entrained pChIs to pause with cortical electrical stimulation29. Compared to optogenetic manipulations20,21,23,38, which depress a subpopulation of ChIs to reduce spike activity, activation of the cortex can ensure that most of the pChIs pause in synchrony29,39 to mimic the synchronised pChI pause observed in behavioural animals26,27. In addition, the pChI pause entrained by cortical stimulation is naturally flanked by endogenous excitations, which may themselves encode reward-relevant information40,41. Thalamic input in particular has a prominent role in inducing multiphasic ChI responses during learning42,43, and is harnessed in Experiment 1 to provide the critical depolarisation required for plasticity, through the effect of the light flash activating the disinhibited tecto-thalamic pathway35,36. Further work is required to combine cortical and thalamic inputs to induce ChI multiphasic activity to fully mimic neuronal activities in freely moving animals and to test their role in synaptic plasticity and learning. In addition, a GABAergic input from the ventral tegmental area (VTA) may also contribute to the formation of the ChI pause in the ventral striatum44, indicating acetylcholine and dopamine system may interplay in a different manner in the ventral striatum than dorsal striatum.
We further demonstrated that changes in pChI activity without phasic dopamine do not induce long-term potentiation at corticostriatal synapses (control group in experiment 1, depol-only group in experiment 2), consistent with others’ findings38. Thus, because phasic dopamine activity reduces at late stages of learning whereas the ChI pause remains intact, the resulting effect of the interplay between ChI activity and dopamine afferents at the striatal level may be dependent on the stage of learning. In addition, manipulating dopamine neurons to fire phasically can only induce synaptic potentiation if it coincides with the pChI pause but not the excitation phase, indicating that the ChI pause is the time window for synaptic potentiation.
We here also addressed how phasic dopamine contributes to synaptic plasticity without using dopamine receptor antagonists, which undesirably also block the effects of tonic dopamine. Indeed, a previous ex vivo study showed that tonic activation of dopamine receptors can reverse the direction of long-term plasticity induced by cortical stimulation45. To avoid blocking the tonic effects of dopamine, our control experiments instead altered the timing of phasic dopamine or omitted dopamine neuron stimulation altogether. We found that even shifting the timing of phasic dopamine activity marginally by a few hundred milliseconds is powerful enough to reverse the direction of the long-term plasticity. Also, when phasic activation of dopamine neurons was omitted, LTD or no change of synaptic efficacy was found. It should be noted that our study focused on mimicking the cell body activity in ChIs and dopamine neurons, as revealed earlier15. However, the release profile of dopamine may not precisely mirror the firing pattern of dopamine neurons due to the potential axonal release driven by ChIs23,24, and should be addressed in future work. Overall, these experiments indicate that the LTP we observed is dependent on phasic dopamine.
Our results also provide further support for the requirement for depolarisation of the target postsynaptic SPNs for long-term potentiation in vivo46 and is consistent with previous ex vivo observations31. This suggests that depolarisation of postsynaptic SPNs may play a critical role in constraining long-term potentiation to synapses on neurons engaged in the action of retrieving rewards. While SPNs in the direct pathway and indirect pathway express distinct sets of dopamine and muscarinic receptors, these neurons may be potentiated to different degrees during learning, which is worth addressing in future studies. In summary, our study provides empirical data supporting the important temporal gating of corticostriatal synaptic plasticity by a physiologically-induced pause response in ChIs and depolarisation of the postsynaptic SPNs.
Methods
All procedures in this study were conducted in accordance with approvals granted by the University of Otago Animal Ethics Committee or in accordance with the UK animals scientific procedures act, 1986 with approval of the University of Bristol ethics committee. A total of 136 male Long-Evan rats for extracellular recording were used, yielding 258 putative spiny projection neurons, and 18 were successfully recorded for the full plasticity recording protocol. The intracellular recording was performed with 105 male Wistar rats, yielding 32 spiny projection neurons recorded for the plasticity protocol.
Surgery
Male Long–Evans rats (250–450 g) or Wistar rats (280– 50 g) were anaesthetised with urethane (1.4–1.9 g/kg i.p.; Biolab Ltd., Auckland, New Zealand). During recording, the level of anaesthesia was monitored by continuous observation of the bandpass filtered EEG signal (0.01–500 Hz). Supplementary urethane was administered via an intraperitoneal catheter at any sign of EEG desynchronisation, indicating a lessening of depth of anaesthesia. The head was fixed in a stereotaxic frame (Narishige, Japan) and core temperature maintained above 36 °C by a homoeothermic blanket and rectal probe (TR-100, Fine Science Tools). All wounds and pressure points were infiltrated with a long-acting local anaesthetic (Bupivacaine, 0.5%).
For monitoring of the electroencephalogram (EEG), a hole was drilled in the skull above the left posterior cortex, and a silver wire electrode placed against the dura overlying the cortex and fixed in placed with dental cement. A flap of bone overlying the cortex was removed to provide access to the recording site in the left medial striatum, and a “well” of dental cement fashioned around the perimeter of the hole. All coordinates are given in millimetres in relation to Bregma and the midline.
Electrical stimulation
To implant a stimulating electrode into the medial agranular motor cortex, a round piece of skull overlying the right hemisphere (centred AP + 2.0 to +2.7 mm and ML −1.6 to −2.0 mm to Bregma) was removed. A concentric (extracellular experiments; Rhodes NEW-100 × 10 mm, USA) or parallel-contact (intracellular experiments, locally manufactured) stimulating electrode was implanted in the medial agranular motor cortex to a depth of 1.6 to 2.4 mm. Stimulating electrodes were connected to constant current electrical stimulators (Isolator-10, Axon Instruments Inc.) Stimulus pulses applied to the cortex were biphasic (0.1–0.2 Hz, 0.1 ms, 300 to 990 µA). For experiments requiring substantia nigra stimulation, the medial contact of a parallel-contact bipolar stimulating electrode was implanted at interaural coordinates AP + 3.4 to +3.6; ML + 1.6; DV 2.1 to 2.3. Substantia nigra stimulation consisted of 50 biphasic pulses (0.5 ms duration) applied at 100 Hz (average current applied for each group 500 to 990 uA).
Visual stimulation
In the extracellular recording experiments, visual stimuli (10 ms duration, 0.2 Hz) were delivered by a white LED (1500 mcd) that was placed 1–2 cm directly in front of the right eye of the animal. The left eye was covered. LED and electrical stimulating electrodes were connected to constant current electrical stimulators (Isolator-10, Axon Instruments Inc.).
Bicuculline injections
The drug-filled pipettes were lowered to 4.0–4.2 mm from the brain surface into the deep layers of the superior colliculus (AP −6.5/ ML + 1.5 mm), and either supported by the IVM micromanipulator or secured with dental cement. Bicuculline (0.01% in saline, 250 nl) was injected into the superior colliculus at a rate of 400 nl/min.
Extracellular recording
Extracellular single-unit recordings were made using 5–15 MΩ micropipettes. Electrodes were filled with 1 M NaCl solution with 2% neurobiotin (SP1120, Vector). Only stable striatal neurons with wide average spike waveform (>1.1 ms), and slow spontaneous firing rate (<0.1 Hz), e.g. Supplementary Fig. 2, were included. Neurons with train spike activity typical of low threshold-spiking (LTS) neurons were also excluded from this study. The spike rate of the recorded SPNs during 30 ms following each cortical stimulation was used as an indication of the strength of corticostriatal synapses. Recordings were made via either a headstage (model HS-2A) connected to an Axoprobe-1A microelectrode amplifier (Axon Instruments Inc California, USA), or a headstage (NL 100 Neurolog) connected to a preamp (NL104), an amplifier (NL106) and a filter (NL125). Signals were amplified and band-pass filtered (0.1–10,000 Hz). All waveform data were digitised at 50 kHz by an A-D interface (1401 Micro 2, CED, UK), and acquired using SPIKE2 software (v6 or v7, CED).
Dopamine neurons were extracellularly recorded and juxtacellularly labelled (AP −5(±0.3) ML + 2(±0.3) DV −7/−8) using glass electrodes as described above. Single-unit data were acquired with an ELC-01MX headstage (NPI electronic), amplified 1000 times, and bandpass filtered at 300 to 5000 Hz (DPA-2FS filter). Depth of anaesthesia was monitored using the electrocorticogram (amplified 2000 times and bandpass filtered at 300 to 1500 Hz) from a stainless-steel screw implanted at AP +2, ML +2. Data were digitised at 25 kHz using a CED Power 1401 mkII.
Intracellular recording
Intracellular recordings were made using 35–130 MΩ micropipettes with 1 M K-acetate internal solution, in some cases containing 3–4% biocytin. For intracellular recording, only stable neurons with a membrane potential more negative than –60 mV that displayed characteristic spontaneous fluctuations in membrane potential (>10 mV amplitude) and action potential firing were included in this study. Current–voltage relations were obtained by injecting hyperpolarising and depolarising current pulses through the micropipette, using an Axoclamp-2B amplifier (Molecular Devices) configured in current-clamp mode. Membrane potential fluctuations were recorded for periods of at least a minute after the cell had stabilised following impalement and at regular intervals of >15 min. All waveform data were digitised at 10 kHz by a Digidata 1200B or a Digidata 1322A (Molecular Devices), displayed using pClamp 8 software (Molecular Devices).
Extracellular recording experimental protocol
After a stable single-unit recording was obtained from a putative spiny projection neuron, cortical stimulation (0.2 Hz) was applied throughout the recording, and the short latency (<30 ms) spike response was measured as the strength of the corticostriatal synaptic input. During the recording, firstly, a baseline of 10 min of spike activity was recorded. The cortical stimulation was then paired with light stimulation for another 5 min. Bicuculline was then injected locally to the deep layers of the superior colliculus. Visual stimulation was paired for another 10 min with the cortical stimulation. In the match group, visual stimulation was applied when the iLFP started to decrease, approximately 500 to 600 ms after cortical stimulation. In the mismatch group, visual stimulation was applied when the iLFP started to increase, approximately 250 ms after cortical stimulation. Therefore, the estimated light-induced phasic dopamine activity occurred either during the ChI pause (Match group) or excitation (Mismatch group, Fig. 1c). In the control group, the visual simulation was not applied after BIC injection. After pairing, the recording of spike responses to cortical stimuli continued for at least another 30 min.
The peristimulus histograms (PSTH) of putative ChIs and SPN spikes were plotted using the cortical stimulation (Figs. 1c, 2a) or the trough of the iLFP (Fig. 3c) as the triggers. Each PSTH represents 60 sweeps of recording and the bin sizes were 50, 1, and 20 ms in Figs. 1c, 2a, and 3c, respectively.
Intracellular recording experimental protocol
After a stable impalement was obtained from a spiny projection neuron, a baseline of 20 min of postsynaptic potentials (PSPs) was recorded before the plasticity-inducing protocol. Postsynaptic potentials were always elicited from the Down state. The transitions between membrane potential “states” were detected online using a locally-constructed functional clamp47, and this used to trigger data acquisition and substantia nigra electrical stimulation, where relevant, early in the ensuing Down or Up state. A delay of 600 ms was imposed between intracellular current injection and substantia nigra stimulation in the dissociated group to ensure that the depolarisation was induced in the up state, and the substantia nigra stimulation occurred primarily in the ensuing down state. Intracellular current injection, when included in the protocol, was set 0.2 nA higher than the threshold for eliciting continuous action potential firing (range 0.5–2.0 nA). Membrane potential fluctuations and current–voltage relations were assessed before and after the plasticity-inducing protocol to ensure that changes in PSPs were not associated with changes in membrane characteristics. The experimental protocols used are illustrated in Fig. 3c.
Histology
At the end of extracellular recording, the putative spiny neurons were actively filled with neurobiotin by a juxtacellular filling protocol48. Briefly, the target cells were “driven” to spike by applying positive current through the recording pipette (up to 12 nA, 250 ms on–off pulses) for up to 15 min. At the end of intracellular recording, putative spiny neurons were filled intracellularly with biocytin by applying depolarising current pulses (0.8–1.5 nA; 3 Hz; 10–15 min) via the recording micropipette.
Vibratome sections (50–60 µm) were processed using standard histological procedures49 and labelled cells were identified by light or fluorescent microscopy. In the extracellular recording experiments, 8 neurons, and in the intracellular recording experiments, 14 neurons, were recovered and verified histologically as spiny projection neurons. Juxtacellularly labelled dopamine neurons were identified as previously described (Dodson et al., 2016) using Cy3-conjugated streptavidin (1:500, Sigma-Aldrich GEPA43001), chicken anti-Tyrosine Hydroxylase primary (1:500, Abcam AB76442) and Brilliant Violet 421-conjugated anti-chicken secondary antibodies (1:500, Jackson ImmunoResearch 703-675-155)50.
Positions of the substantia nigra electrodes for intracellular recordings were determined in unstained or cresyl-violet stained sections to be within 500 µm of the dopamine cell layer of the substantia nigra pars compacta. There were no systematic differences in electrode positions between groups. In groups where SN stimulation was not applied, electrodes were still fitted during surgery to control for the release of dopamine that may accompany acute electrode implantation51.
Data analysis
Data were analysed offline using built-in functions of Axograph 4.9 software and SPIKE2 v6 or v7. Statistical tests on data from single cells as well as on group data were performed in SPSS and Prism.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Date availability
Raw data for Figs. 1 and 2 are available if required. Source data for figures are provided with the paper. Source data are provided with this paper.
References
Schultz, W., Dayan, P. & Montague, P. R. A neural substrate of prediction and reward. Science 275, 1593–1599 (1997).
Wise, R. A. Dopamine, learning and motivation. Nat. Rev. Neurosci. 5, 483–494 (2004).
Surmeier, D. J., Plotkin, J. & Shen, W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr. Opin. Neurobiol. 19, 621–628 (2009).
Reynolds, J. N., Hyland, B. I. & Wickens, J. R. A cellular mechanism of reward-related learning. Nature 413, 67–70 (2001).
Reynolds, J. N. & Wickens, J. R. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 15, 507–521 (2002).
Fisher, S. D. et al. Reinforcement determines the timing dependence of corticostriatal synaptic plasticity in vivo. Nat. Commun. 8, 334 (2017).
Hamid, A. A. et al. Mesolimbic dopamine signals the value of work. Nat. Neurosci. 19, 117–126 (2016).
Howe, M. W. & Dombeck, D. A. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535, 505–510 (2016).
Coddington, L. T. & Dudman, J. T. The timing of action determines reward prediction signals in identified midbrain dopamine neurons. Nat. Neurosci. 21, 1563–1573 (2018).
Howe, M. et al. Coordination of rapid cholinergic and dopaminergic signaling in striatum during spontaneous movement. Elife 8, e44903 (2019).
Howard, C. D., Li, H., Geddes, C. E. & Jin, X. Dynamic nigrostriatal dopamine biases action selection. Neuron 93, 1436–1450 e1438 (2017).
Engelhard, B. et al. Specialized coding of sensory, motor and cognitive variables in VTA dopamine neurons. Nature 570, 509–513 (2019).
Cragg, S. J. Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci. 29, 125–131 (2006).
Joshua, M., Adler, A., Mitelman, R., Vaadia, E. & Bergman, H. Midbrain dopaminergic neurons and striatal cholinergic interneurons encode the difference between reward and aversive events at different epochs of probabilistic classical conditioning trials. J. Neurosci. 28, 11673–11684 (2008).
Morris, G., Arkadir, D., Nevet, A., Vaadia, E. & Bergman, H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43, 133–143 (2004).
Cox, J. & Witten, I. B. Striatal circuits for reward learning and decision-making. Nat. Rev. Neurosci. 20, 482–494 (2019).
Deffains, M. & Bergman, H. Striatal cholinergic interneurons and cortico-striatal synaptic plasticity in health and disease. Mov. Disord. 30, 1014–1025 (2015).
Calabresi, P., Picconi, B., Tozzi, A. & Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219 (2007).
Goldberg, J. A., Ding, J. B. & Surmeier, D. J. Muscarinic modulation of striatal function and circuitry. Handb. Exp. Pharmacol. 208, 223–241 (2012).
Witten, I. B. et al. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science 330, 1677–1681 (2010).
Nelson, A. B. et al. Striatal cholinergic interneurons Drive GABA release from dopamine terminals. Neuron 82, 63–70 (2014).
Rice, M. E. & Cragg, S. J. Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 7, 583–584 (2004).
Threlfell, S. et al. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64 (2012).
Cachope, R. et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2, 33–41 (2012).
Wang, L. et al. Modulation of dopamine release in the striatum by physiologically relevant levels of nicotine. Nat. Commun. 5, 3925 (2014).
Aosaki, T. et al. Responses of tonically active neurons in the primate’s striatum undergo systematic changes during behavioral sensorimotor conditioning. J. Neurosci. 14, 3969–3984 (1994).
Apicella, P., Deffains, M., Ravel, S. & Legallet, E. Tonically active neurons in the striatum differentiate between delivery and omission of expected reward in a probabilistic task context. Eur. J. Neurosci. 30, 515–526 (2009).
Ravel, S., Legallet, E. & Apicella, P. Responses of tonically active neurons in the monkey striatum discriminate between motivationally opposing stimuli. J. Neurosci. 23, 8489–8497 (2003).
Zhang, Y. F., Reynolds, J. N. J. & Cragg, S. J. Pauses in cholinergic interneuron activity are driven by excitatory input and delayed rectification, with dopamine modulation. Neuron 98, 918–925 e913 (2018).
Matsuda, W. et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 29, 444–453 (2009).
Wickens, J. R., Begg, A. J. & Arbuthnott, G. W. Dopamine reverses the depression of rat corticostriatal synapses which normally follows high-frequency stimulation of cortex in vitro. Neuroscience 70, 1–5 (1996).
Ryan, L. J., Tepper, J. M., Young, S. J. & Groves, P. M. Frontal cortex stimulation evoked neostriatal potentials in rats: intracellular and extracellular analysis. Brain Res. Bull. 17, 751–758 (1986).
Comoli, E. et al. A direct projection from superior colliculus to substantia nigra for detecting salient visual events. Nat. Neurosci. 6, 974–980 (2003).
Dommett, E. et al. How visual stimuli activate dopaminergic neurons at short latency. Science 307, 1476–1479 (2005).
Schulz, J. M. et al. Short-latency activation of striatal spiny neurons via subcortical visual pathways. J. Neurosci. 29, 6336–6347 (2009).
Redgrave, P., Vautrelle, N. & Reynolds, J. N. Functional properties of the basal ganglia’s re-entrant loop architecture: selection and reinforcement. Neuroscience 198, 138–151 (2011).
Stern, E. A., Jaeger, D. & Wilson, C. J. Membrane potential synchrony of simultaneously recorded striatal spiny neurons in vivo. Nature 394, 475–478 (1998).
Lee, J., Finkelstein, J., Choi, J. Y. & Witten, I. B. Linking cholinergic interneurons, synaptic plasticity, and behavior during the extinction of a cocaine-context association. Neuron 90, 1071–1085 (2016).
Reynolds, J. N. J. & Wickens, J. R. The corticostriatal input to giant aspiny interneurons in the rat: a candidate pathway for synchronising the response to reward-related cues. Brain Res. 1011, 115–128 (2004).
Apicella, P., Ravel, S., Deffains, M. & Legallet, E. The role of striatal tonically active neurons in reward prediction error signaling during instrumental task performance. J. Neurosci. 31, 1507–1515 (2011).
Goldberg, J. A. & Reynolds, J. N. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience 198, 27–43 (2011).
Matsumoto, N., Minamimoto, T., Graybiel, A. M. & Kimura, M. Neurons in the thalamic CM-Pf complex supply striatal neurons with information about behaviorally significant sensory events. J. Neurophysiol. 85, 960–976 (2001).
Bradfield, L. A., Bertran-Gonzalez, J., Chieng, B. & Balleine, B. W. The thalamostriatal pathway and cholinergic control of goal-directed action: interlacing new with existing learning in the striatum. Neuron 79, 153–166 (2013).
Brown, M. T. et al. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492, 452–456 (2012).
Kerr, J. N. D. & Wickens, J. R. Dopamine D-1/D-5 receptor activation is required for long-term potentiation in the rat neostriatum in vitro. J. Neurophysiol. 85, 117–124 (2001).
Charpier, S. & Deniau, J. M. In vivo activity-dependent plasticity at cortico-striatal connections: evidence for physiological long-term potentiation. Proc. Natl Acad. Sci. USA 94, 7036–7040 (1997).
Reynolds, J. N. J. & Wickens, J. R. A state-dependent trigger for electrophysiological recording at predetermined membrane potentials. J. Neurosci. Methods 131, 111–119 (2003).
Pinault, D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. J. Neurosci. Methods 65, 113–136 (1996).
Horikawa, K. & Armstrong, W. E. A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J. Neurosci. Methods 25, 1–11 (1988).
Dodson, P. D. et al. Representation of spontaneous movement by dopaminergic neurons is cell-type selective and disrupted in parkinsonism. Proc. Natl Acad. Sci. 113, E2180–E2188 (2016).
Moghaddam, B., Adams, B., Verma, A. & Daly, D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 17, 2921–2927 (1997).
Acknowledgements
Our thanks to Annabel Kean and Koreen Clements for technical assistance, and to Jan Schulz, Peter Redgrave and Gordon Arbuthnott for helpful discussions that have shaped this manuscript. This work was supported by grants from the PhD scholarship, Department of Anatomy, University Otago (to Y-FZ), Marsden Fund of the Royal Society of NZ (to JNJR, and JRW/JNJR), Lottery Health Research (JNJR/JRW) and the Neurological Foundation of New Zealand (MJO/JNJR).
Author information
Authors and Affiliations
Contributions
Conceptualisation, Y.-F.Z., J.R.W., and J.N.J.R.; Data curation, Y.-F.Z. and J.N.J.R.; Investigation, Y.-F.Z., extracellular recordings, J.N.J.R. intracellular recordings: R.A., P.D.D. dopamine neuron recordings; Formal analysis, Y.-F.Z., extracellular recordings, J.N.J.R intracellular recordings, R.A., P.D.D. dopamine neuron recordings; Methodology, Y.-F. Z., J.R.W., M.J.O. and J.N.J.R.; Funding acquisition and resources, J.R.W. and J.N.J.R.; Writing—original draft, Y.-F. Z. and J.N.J.R.; Writing—review & editing, Y.-F.Z., S.D.F., M.J.O., J.R.W., R.A., P.D.D., and J.N.J.R.; Supervision, J.N.J.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Paul Apicella and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reynolds, J.N.J., Avvisati, R., Dodson, P.D. et al. Coincidence of cholinergic pauses, dopaminergic activation and depolarisation of spiny projection neurons drives synaptic plasticity in the striatum. Nat Commun 13, 1296 (2022). https://doi.org/10.1038/s41467-022-28950-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28950-0
This article is cited by
-
Ventral tegmental area dopamine projections to the hippocampus trigger long-term potentiation and contextual learning
Nature Communications (2024)
-
Adaptive control of synaptic plasticity integrates micro- and macroscopic network function
Neuropsychopharmacology (2023)
-
Acetylcholine waves and dopamine release in the striatum
Nature Communications (2023)
-
Intrinsic dopamine and acetylcholine dynamics in the striatum of mice
Nature (2023)
-
Spontaneous behaviour is shaped by dopamine in two ways
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
