
Abstract
Lytic polysaccharide monooxygenase (LPMO) supports biomass hydrolysis by increasing saccharification efficiency and rate. Recent studies demonstrate that H2O2 rather than O2 is the cosubstrate of the LPMO-catalyzed depolymerization of polysaccharides. Some studies have questioned the physiological relevance of the H2O2-based mechanism for plant cell wall degradation. This study reports the localized and time-resolved determination of LPMO activity on poplar wood cell walls by measuring the H2O2 concentration in their vicinity with a piezo-controlled H2O2 microsensor. The investigated Neurospora crassa LPMO binds to the inner cell wall layer and consumes enzymatically generated H2O2. The results point towards a high catalytic efficiency of LPMO at a low H2O2 concentration that auxiliary oxidoreductases in fungal secretomes can easily generate. Measurements with a glucose microbiosensor additionally demonstrate that LPMO promotes cellobiohydrolase activity on wood cell walls and plays a synergistic role in the fungal extracellular catabolism and in industrial biomass degradation.
Similar content being viewed by others

Introduction
The discovery of lytic polysaccharide monooxygenase (LPMO) featuring a surface exposed active-site with a type-2 copper center, has changed the perception of fungal lignocellulose degradation1,2,3,4. LPMOs employ an oxidative rather than a hydrolytic mechanism to cleave glycosidic bonds in cellulose3,5, hemicelluloses6, chitin2, starch7, xylan8 and soluble cello-oligosaccharides9 (carbohydrate-active enzyme CAZy auxiliary activities AA9-12, AA13-17). LPMOs have been widely identified in fungi8, bacteria2 and others. The research on LPMOs is driven by their ability to promote biomass saccharification10,11,12. LPMO has also been found to play an opposing role in the pathogen-defense mechanism of plants13,14. The elucidation of LPMO’s reaction mechanism, its activity on natural substrates and its interaction with other enzymes is a prerequisite for advanced applications in plant biomass hydrolysis like the selective depolymerization in biorefineries and for understanding its role in physiological processes. Previous work demonstrated that each LPMO, irrespective of its origin, needs an electron donor to activate the copper center to prime the catalytic cycle15. The flavocytochrome cellobiose dehydrogenase (CDH) is the proposed native reductase of LPMO16,17. Once activated, the copper center utilizes an oxygen species as cosubstrate to hydroxylate the C1 or the C4 atom of the polysaccharide substrate, thereby breaking the glycosidic bond9,18,19. The nature of the cosubstrate has initially been assumed to be O2, but research in the last years brought forth ample evidence for H2O2 as actual cosubstrate20,21,22,23,24,25. The catalytic efficiency of LPMO for polysaccharide depolymerization is much higher when adding H2O2 compared to using the preliminary reduction of O2 to H2O2 either directly by the added electron donor (reductant) or via an oxygen uncoupling mechanism of the reduced, non-substrate bound LPMO20,26. The reaction rate of the activated LPMO9A from Trichoderma reesei (TrLPMO9A) and H2O2 was three orders of magnitude faster than for O223. The reported kcat/KM values of bacterial and fungal LPMOs for H2O2 are in the order of 106 M−1 s−1 24,27, which is comparable to catalytic efficiencies of fungal peroxygenases28 and peroxidases29. Conversely, the catalytic efficiency of a fungal LPMO for O2-dependent substrate oxidation was measured to be 103 M−1 s−1 22. In an industrial setting, saccharification rates and yields for treatment of both pure cellulose and birch samples increased when H2O2 was supplied for LPMO and cellulases21. While aiming for higher efficiency of cellulose saccharification, H2O2-driven LPMO catalysis has not yet been studied at the structural level of wood cells which form the natural environment of fungal hyphae and their secreted enzymes. To measure the peroxygenase activity of LPMO on plant cell walls under near-natural conditions and in the presence of cellulases is an essential step to understand the enzyme’s natural function.
The study of LPMO’s reaction kinetics and interaction with other enzymes is hampered by the necessity of an analytical method that overcomes the experimental difficulties associated with the investigation of enzymes bound to heterogeneous and structurally complex biopolymers in plant cell walls30,31. In the secondary cell wall, the main target of fungal exoenzymes, the cellulose microfibrils are surrounded by hemicellulose and inter-connected lignin networks, causing the recalcitrance of lignocellulosic biomass towards microbial deconstruction32. LPMOs are active on cellulose and hemicelluloses and are a substantial constituent of fungal secretomes6. Several studies report that LPMO can either promote or impede cellobiohydrolase activity depending on the studied enzymes and substrates12,33,34. A combination of TrCel6A and TrLPMO9A showed a 2–2.5-fold increase in the degradation of amorphous cellulose compared to the same dosage of individual enzyme but showed negligible effect on crystalline cellulose33. The addition of C1-oxidizing LPMO from Thielavia terrestris increased the glucose production from bacterial cellulose when administered with TrCel6A or TrCel7B, however, it decreased glucose production when administered with TrCel7A12. LPMOs alter the physical accessibility and chemical structure of cellulose chains and can promote the activity of cellobiohydrolases24,25. This effect is of great significance since cellobiohydrolases are the most abundant enzymes in the secretomes of most wood-decaying fungi35,36.
Although LPMO activity can be measured by different assays, no currently available method is able to detect its activity at the microscale when bound to a natural substrate. In this work a H2O2 microsensor was developed and positioned closely above a poplar wood microtome slice by using a scanning electrochemical microscopy (SECM) platform to investigate LPMO’s peroxygenase activity on the surface of wood cell walls in a physiologically relevant environment. Since a microsensor is barely limited by mass transfer and only marginally depletes the analyte, the set-up allows the real-time detection of H2O2 consumption by LPMO bound onto wood cell walls for the first time. Cellobiohydrolases from T. reesei and a CDH variant from Crassicarpon hotsonii (syn. Myriococcum thermophilum) that possesses a 30-fold higher oxygen turnover than the native CDH were used to hydrolyze cellulose in wood cell walls and generate H2O2 in situ for Neurospora crassa LPMO9C37 (Fig. 1). The enhanced H2O2 production rate of the ChCDH variant enabled us to shorten the experimental time (≤ 4 h) to maintain the microsensors’ performance and reduce positioning errors from substrate swelling. A glucose-detecting microbiosensor was additionally used to study the effect of H2O2-driven LPMO activity on the activity of cell wall-bound cellobiohydrolases.

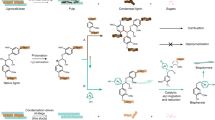
In this study, fungal cellobiohydrolases (light brown, model based on PDB IDs: 7cel and 5x34) and ChCDH (yellow, PDB ID: 4qi5) are used to imitate fungal H2O2 production on poplar wood cell walls. The H2O2 is consumed by NcLPMO9C (cyan, PDB ID: 4d7u) after priming by its auxiliary electron donor NcCDH (magenta, PDB ID: 4qi7). Oxygen is provided from air.
Results
Positioning of a micro(bio)sensor
An SECM platform is composed of a stepper motor with a piezo module, a lock-in amplifier, a potentiostat, an inverted fluorescence microscope and a positioning microscope and the parts are placed in two adjacent Faraday cages (Fig. 2a and Supplementary Fig. 1). A shear-force based SECM mode was employed to position a micro(bio)sensor in close vicinity to the cell walls in cross-section ultramicrotome slices of poplar wood. The resonance frequency of a fixed microsensors (e.g., ~396 kHz) was determined by searching the magnitude difference of the vibrating tip operation in air and in solution (Fig. 2b).

a Schematic representation of the instruments combined in the SECM platform. b Frequency spectra were acquired using the shear-force mode and two piezo-crystals to oscillate the microsensor and measure its attenuation in air (blue) and in solution (black). The red line represents the magnitude difference which indicates 396 kHz as the preferable resonance frequency of the used microsensor. The resonance frequencies of all used micro(bio)sensors were in the range of 340–416 kHz. c The approach curve of a microsensor shows the magnitude changes at the resonance frequency with decreasing distance from the poplar wood surface. d Image of an approached microsensor laterally near a poplar wood slice (in buffer solution) obtained using the inverted microscope. e Image of a positioned microsensor above a poplar wood slice (in buffer solution) from the positioning video microscope. f A microsensor with the mounted piezo plates is fixed on the black cantilever of the positioning unit. The view in red rectangle is zoomed in and shown in g. g Light microscopic images of a Pt ultramicroelectrode. High-magnification images of a middle and a tip segment were shown in the bottom right corner. Scale bars, 50 µm (d); 50 µm (e); 10 µm (g).
The microsensor was moved towards the poplar wood slice surface, and the approaching stopped once the change of the shear-force vibration magnitude overcame 5%, which occurred at a distance of ~300 nm above the sample surface (Fig. 2c, d). Finally, the microsensor was retracted to 25 µm above the surface of the poplar wood cells in all experiments (Fig. 2d, e).
Performance of the H2O2 microsensor
A platinum wire was embedded in a quartz capillary by laser-assisted electrode pulling and resulted in ultramicroelectrodes with a tip length of 1.0–1.5 cm, an electrode diameter between 1–2 µm, and an outer diameter of 20 ± 5 µm (Fig. 2f, g). With an electrodeposition method, the Pt microelectrode was modified by a Prussian blue layer (Fig. 3a). The H2O2 microsensors were characterized by a pair of redox waves (Supplementary Fig. 2) consistent with the redox cycle between Prussian blue and Prussian white38,39. The scanning electron microscope images showed the Pt ultramicroelectrode is covered with Prussian blue particles (Fig. 3b, c). Cyclic voltammograms show that H2O2 reduction reaction on a Prussian blue-modified Pt ultramicroelectrode starts from 0.1 V vs. Ag|AgCl during the cathodic scan (Supplementary Fig. 3). The Prussian blue-modified H2O2 microsensor shows a negligible current for oxygen reduction reaction compared to a bare Pt microelectrode which is a necessary feature to perform measurements under natural, air-saturated conditions (Supplementary Figs. 4 and 5). The high selectivity of the electrocatalyst for H2O2 reduction is consistent with previous reports40 and ensures an extremely low background signal allowing the selective and sensitive detection of H2O2 in presence of O2. The amperometric response of reducing various concentrations of H2O2 stabilizes after 40 s (Fig. 3d). Steady-state currents of H2O2 reduction were calculated as mean values from the time interval between 40 and 50 s and used to plot a calibration curve (Fig. 3e). A linear correlation was found in the measured range between 25–200 µM H2O2 and a sensitivity of 0.078 pA µM−1 µm−2 was calculated. The sensitivity of the shown H2O2 microsensor was re-examined after four hours of a typical experiment to be 0.066 pA µM−1 µm−2. This 14.3% reduction of the sensitivity is acceptable but meanwhile shows the time limit of highly sensitive, real-time measurements of H2O2 microsensors.

a Schematic principle of Prussian blue based H2O2 microsensor. b, c are scanning electron microscope images of a Pt ultramicroelectrode and the Prussian blue modified Pt ultramicroelectrode. Scale bars, 10 µm (b); 10 µm (c). d Amperometric response of a H2O2 microsensor to 0 µM (black,), 25 µM (gray), 50 µM (green), 100 µM (blue), 150 µM (magenta) and 200 µM (red) H2O2. e Calibration plots of the H2O2 microsensors before (solid line) and after 4-h experimental use (dashed line). The currents were extracted from the mean values between 40 s and 50 s of the measurements. Data in e are shown as means values, and error bars show SD (n = 3, independent experiments).
Localized formation and detection of H2O2
A cell wall-associated enzymatic production of H2O2 mimics the fungal degradation mechanism and provides a more realistic scenario than external cosubstrate addition when studying LPMO activity. The combination of purified T. reesei cellobiohydrolases and a C. hotsonii CDH variant capable of binding to poplar wood slices by their carbohydrate-binding modules were used to produce H2O2 in a catalytic cascade reaction. The cellobiohydrolases activity converts cellulose into cellobiose which is oxidized by CDH using O2 as an electron acceptor. The C. hotsonii CDH variant with a 30-fold higher oxygen turnover than the wild type37,41 was used to obtain H2O2 concentrations that can be reliably quantified by a microsensor within the experimental time limits. Upon adding both enzymes, the localized H2O2 concentration in the close vicinity of the poplar wood cell walls increased over 120 min and reached 106.1 ± 6.7 µM at the end (Fig. 4a). The observed, almost linear increase demonstrates a relatively stable enzymatic activity during the experiment. No H2O2 production was detected in the control experiment without ChCDH. An experiment in which ChCDH was substituted by glucose oxidase (GOX) and β-glucosidase resulted in an almost two-fold higher H2O2 concentration of 208.5 ± 6.2 µM, which demonstrates that the availability of the substrates cellulose and cellobiose were not rate-limiting for the CDH catalyzed reaction. These results verify that the microsensor is able to conduct real-time measurements of H2O2 in the close vicinity of poplar wood cell walls.

a The graph shows time courses of H2O2 formation by 2 mg mL−1 cellobiohydrolases and 1 µM C. hotsonii CDH variant (filled circles) or 2 mg mL−1 cellobiohydrolases and 1 mg mL−1 β-glucosidase in combination with 1 µM GOX (filled triangles) during hydrolysis of poplar wood cell walls in the absence of LPMO. In the control experiment (empty squares), only 2 mg mL−1 cellobiohydrolase has been applied. b The change of O2 (gray circles) and H2O2 concentration (black circle) in the vicinity of wood cell walls before and after LPMO catalysis. The additions of 1 µM LPMO9C and 1.0 µM N. crassa CDHIIA are indicated by a black arrow. c Time courses of H2O2 consumption by 0.5 µM (black circle), 1.0 µM (red circle), and 3.0 µM (blue circle) LPMO9C in combination with 1.0 µM N. crassa CDH. Their additions after 180 min are indicated by a gray arrow. In the experiments shown in b, c, 2 mg mL−1 cellobiohydrolases and 1 µM C. hotsonii CDH variant are applied since the beginning to produce H2O2. All experiments were conducted in a 50 mM sodium acetate buffer, pH 5.5 at ∼20 °C. The reference experiment without addition of LPMO is given in Fig. 5a (black circles). Data in a are shown as means values, and error bars show SD (n = 3, independent experiments). Data in c are shown as means values, and error bars show SD (n = 2, independent experiments).
Detection of local H2O2 consumption by LPMO
The developed microsensor approach is currently the only available method to measure the peroxygenase activity of LPMO on an intact plant cell wall sample. The recently published turbidimetric42 and photometric assays24 depend on purified and disperse substrates. To study LPMO activity on microtome slices of poplar wood as a realistic approximation of the natural conditions, N. crassa LPMO9C and NcCDHIIA were delivered to the poplar wood slice surface using a microsyringe with the aid of a video microscope. N. crassa CDH (not the H2O2-producing C. hotsonii CDH variant) was used as an electron donor because it is the natural electron donor for N. crassa LPMOs15,43. It also has a carbohydrate-binding module that ensures its binding to the cell walls. The NcCDH was injected 3 min after LPMO9C to allow the LPMO binding to the cell walls. Before adding LPMO and NcCDH, the H2O2 production rate was similar to the reaction rate in the reference experiment (Fig. 4a) with 0.85 vs. 0.88 µM min−1, respectively. Immediately after the addition, the H2O2 concentration decreased quickly and then leveled off after ~30 min; however, the O2 concentration in the experiment was only reduced by the action of C. hotsonii CDH, but not by the addition of N. crassa LPMO or CDH (Fig. 4b). This result that LPMO does not consume any measurable amount of O2 has been observed in at least three independent experiments and shows the preference of LPMO for H2O2 in the presence of O2.
The stop of observable H2O2 consumption can have three different reasons: (i) reaching an equilibrium between H2O2 formation and consumption, (ii) a slowdown of LPMO activity due to substrate depletion, and (iii) deactivation of LPMO because of the high H2O2 concentration. The injection of three different LPMO concentrations (Fig. 4c) showed that it is not a depletion of substrate available to LPMO but most likely a combination of deactivation and reaching a kinetic equilibrium. This is evident from the increase in H2O2 concentration from 210 to 240 min after adding 0.5 µM LPMO and the stagnation in this time when 3 µM LPMO was added. The high accumulated H2O2 concentration was necessary for the detection by the developed microsensors. In nature, such high H2O2 concentrations are unlikely because of the lower production rate and the continuous consumption by peroxygenases, peroxidases and catalases44. Previous studies have found that the oxidation of uncoupled LPMOs with H2O2 inactivates the enzyme quickly20,45. The structural complexity of wood cell walls resulting in lower accessibility of binding sites for LPMO could intensify their oxidative deactivation. The activity of N. crassa LPMO9C on poplar wood with H2O2 can be estimated from the initial H2O2 consumption rates, which were 3.2–16.7 µM min−1 depending on the added LPMO concentration (0.5–3 µM). The calculation of a specific activity or turnover number is difficult, since from the added LPMO molecules, only a fraction was bound to its substrate and therefore active. The lowest estimate for a turnover number when considering all added LPMO molecules active was 0.1 s−1 calculated from the data presented in Fig. 4c. These values are still six times higher than the reported turnover numbers on amorphous cellulose with O2 as a cosubstrate22,46.
Excessive H2O2 deactivates LPMO
The previous experiments have shown the LPMO can consume the accumulated H2O2 in the surroundings of wood cell walls at a high rate. An interesting question is the actual H2O2 concentration when the LPMO is present at the very beginning of the experiment. Will H2O2 accumulate, or will LPMO consume it and keep it at a low concentration that is not harmful to the enzymes and possibly the fungus?
To answer this question, 1 µM N. crassa LPMO9C and 0.5 µM CDHIIA were simultaneously applied with the H2O2 producing enzymes. In the initial 50 min of the reaction, H2O2 was not detected in the vicinity of the wood cell walls. In the following 70 min (50–120 min), trace amounts of H2O2 were detected (Fig. 5a). These concentrations were lower than the limit of quantitation of the used microsensors (~10 µM), making them less accurate. However, a slow accumulation of H2O2 was indeed observed, which could be the reason for increasing LPMO deactivation. After 120 min, the H2O2 concentration increased at an approximately equal rate as the control reaction without LPMO. At this point, we assumed that nearly all the LPMO enzymes were deactivated and ceased using H2O2. Generally, during the first 120 min, remarkably little H2O2 was detected in the surroundings of the poplar wood cell walls. The presence of LPMO9C therefore efficiently suppressed H2O2 accumulation by using it for catalysis. Based on the total produced H2O2, the total turnover number of LPMO during the first 120 min was calculated to be 103 ± 2. The results also indicated that LPMO can help maintain a very low H2O2 concentration under physiological conditions, which cannot be easily detected and will significantly reduce enzyme deactivation of hyphal damage. The rapid inactivation of LPMOs in the presence of a high H2O2 concentration has been reported in previous studies45. We propose that under natural conditions and even lower physiological H2O2 concentrations produced by fungi the total turnover number of LPMO will be much higher than previously suggested22. To verify the activity of cellobiohydrolases and LPMO, a glucose microbiosensor was employed to quantify the released cellobiose and cello-oligosaccharides in both reactions (Fig. 5b). In both experiments, the activity of the cellobiohydrolases continuously increased the glucose concentration, but the presence of LPMO boosted the amount of reaction product by 19.6% after 120 min. The synergy of the enzymes became less obvious at longer incubation periods which coincided with the deactivation of LPMO after 120 min.

a Time courses of H2O2 formation on wood cell walls in the presence (blue circles) and absence (black circles) of NcLPMO9C while loading 10 mg mL−1 cellobiohydrolases and 1 µM ChCDH in 50 mM sodium acetate buffer, pH 5.5, at 20 °C. b Time courses of glucose formation on wood cell walls in the presence (blue circles) and absence (black circles) of NcLPMO9C while loading 10 mg mL−1 cellobiohydrolases and 2 mg mL−1 β-glucosidase in 50 mM potassium phosphate buffer, pH 6.0, at 20 °C. 1 µM NcLPMO9C with 0.5 µM NcCDHIIA are used in both experiments. Data in a are shown as mean values, and error bars show SD (n = 3, independent experiments), in b data of single control measurements are presented.
Localization of LPMO and CDH on cell walls
To investigate their binding onto the wood cell walls during H2O2-driven catalysis, LPMO and NcCDH were tagged with different fluorescent moieties to check their selective localization on wood cell walls. Fig. 6a, b showed that both enzymes were mainly bound to the S3 secondary cell walls that are rich in cellulose. The unspecific binding of NcCDH was found on the middle lamella and cell corners regions (Fig. 6a). The brightfield image showed the solid cell walls (Fig. 6c). The overlay of both fluorescent channels and the lignin autofluorescence detected between 415 and 550 nm showed abundant overlaps of enzyme binding sites on the cell walls (Fig. 6d). This implied a frequent interaction between these two enzymes during catalysis, especially when LPMO was reduced by NcCDH, which increased the binding affinity of the LPMO47 (Supplementary Fig. 6).

Fluorescence images show the binding of N. crassa CDHIIA and LPMO9C onto poplar wood cell walls. a Fluorescence from DyLight D550-labeled CDH, and b fluorescence from DyLight D633-labeled LPMO. c Image from brightfield microscopy shows the wood cell walls. d An overlay of both fluorescence channels shows the colocalization of CDH and LPMO. Picture size = measurement area = 70 × 70 μm.
LPMO promotes cellobiohydrolase activity
For this experiment, poplar wood sections were pretreated with LPMO9C to modify the cell walls before adding T. reesei cellobiohydrolase acting from the reducing-end (Cel7A) or the nonreducing-end (Cel6A) of a cellulose chain. By adding β-glucosidase, glucose was obtained as the final hydrolysis product, and a glucose microbiosensor was used to assess the cellobiohydrolase activity on poplar wood cell walls. The microbiosensors were calibrated by fitting the steady-state currents vs. the glucose concentrations (Supplementary Fig. 7). The collected data was considered only if the glucose microbiosensors retained a final sensitivity >75% of the initial value after completion of the measurement (Supplementary Fig. 8). The glucose production measured on the LPMO-pretreated poplar wood slices after 30 min increased by 40 and 44% for TrCel6A and TrCel7A, respectively, compared to the untreated samples (Fig. 7). The increased turnover of both cellobiohydrolases after LPMO pretreatment can be either attributed to the formation of additional free ends on cellulose, acting as starting sites for cellobiohydrolases, or the removal of hemicelluloses, which may improve the accessibility of cellulose fibrils.

Glucose production by TrCel6A (a) and TrCel7A (b) while degrading the untreated (black circles) and pretreated (blue triangles) poplar wood slices by NcLPMO9C with its reductant and H2O2 as cosubstrate. All reactions are conducted in a 50 mM potassium phosphate buffer, pH 6.0 at 20 °C. Data are shown as mean values and error bars show SD (n = 3, independent experiments).
Discussion
Ultramicroelectrodes used as micro(bio)sensors have been used previously in SECM to measure the release or consumption of neurotransmitters, H2O2, or glucose from live cells48 or bacteria49. Enzymatic degradation of woody biomass essentially occurs at the wood cell walls. We used the SECM set-up for the positioning of the micro(bio)sensors in a fixed, close position to the wood cell wall to monitor temporal changes in the H2O2 or glucose concentration while loading with various oxidoreductases under experimental conditions closely resembling the natural environment of fungal biomass breakdown.
A Prussian blue thin film endowed the H2O2 microsensor high sensitivity and selectivity when operating in a complex reaction mixture containing several enzymes and other compounds. The results from the control reaction in Fig. 4a verified that none of the matrix components interfered with the measured signals and the measured currents accurately reflect the H2O2 concentration during enzymatic degradation of wood cell walls. Additionally, the microsensor methodology avoids the interfering side reactions from the reductants and oxidants used for other LPMO activity assay methods24,50.
In nature, H2O2 is used by both brown-rot and white-rot fungi to degrade woody biomass through enzymatic and nonenzymatic reactions. Oxidases and also dehydrogenases from the glucose–methanol–choline (GMC) oxidoreductase superfamily have been identified as sources of H2O2, e.g., not only alcohol oxidase from Gloeophyllum trabeum51 or aryl-alcohol oxidase from Pleurotus eryngii52 but also H2O2-producing copper enzymes such as glyoxal oxidase from basidiomycetes53. In this study, an enzyme cocktail containing glycoside hydrolases from T. ressei and a C. hotsonii CDH that can simultaneously degrade wood cell walls and produce H2O2 was applied to mimic the complicated H2O2-producing processes during woody biomass degradation by fungi in nature. Our results clearly showed that N. crassa LPMO9C binds to poplar wood cell walls and consumes H2O2. Although the tip diameter (ca. 1.5 mm) of the used oxygen sensor was much larger than that of the H2O2 microsensor, the close distance from the poplar wood slice surface ensured a fast response. Since no change in the O2 concentration after LPMO addition was observed, this supports the conclusion that LPMO uses H2O2 as cosubstrate even in the presence of O220. The time-resolved measurement of H2O2 and O2 concentrations during LPMO catalysis revealed that LPMO uses H2O2 as cosubstrate and does not consume the present O2 when bound to wood cell walls.
The H2O2-driven LPMO catalysis was observed for more than 2 h in a near-natural environment, but eventually, the high amount of H2O2 produced by the auxiliary enzymes in the experimental set-up leads to LPMO inactivation. The H2O2-producing enyzmes are more stable towards their product than LPMO and are active even after accumulating H2O2 for up to 4 h in the experiments. The production of H2O2 by CDH under natural conditions is regulated by many factors, such as the availability of phenol derived electron acceptors (which reduce the oxygen turnover), the pH which has a strong effect not only on the catalytic activity but also the electron transfer to LPMO via CDH’s cytochrome domain54. The presence of organic acids is an important factor that can therefore not only modulate the production and stability of H2O2 but has also shown to have a modulating effect on LPMO activity55.
Our results demonstrate that LPMOs secreted by wood-degrading fungi scavenge H2O2 present in their surroundings. Although we observed the deactivation of LPMOs in the presence of H2O2, the peroxygenase activity was maintained for up to 120 min in a set-up with a high concentration of H2O2-generating enzymes. When fungi degrade woody biomass, different extracellular peroxidases (e.g., manganese peroxidases; catalytic efficiency (kcat/KM H2O2: 106–107 M−1 s−1) maintain the H2O2 concentration at a relatively low level in the surroundings of wood cell walls56,57. Under these conditions, LPMO’s peroxygenase activity should be sustained much longer than observed in our experiments.
In this experimental set-up, the precise amount of enzyme and substrate concentration in the interfacial area (infinite diffusion hemisphere of the microsensor could not be quantified). Therefore, kinetic constants of LPMO were only reported as estimates. The minimum estimate of the LPMO turnover number for its polymeric substrate (0.1 s−1) is much lower than the kcat for the oxidation of soluble cellopentaose by N. crassa LPMO9C (124 s−1)58 and or for the oxidation of insoluble chitin by a bacterial LPMO (5.6 s−1)59. It is worth noting that the enzyme concentrations in our calculation are grossly overestimated because we considered the total added enzyme instead of the bound enzymes on the wood cell walls. Nevertheless, the H2O2 microsensor is excellent in measuring the peroxygenase activity of various LPMOs on the surface of different solid substrates, as well as other H2O2-consuming or H2O2-producing fungal enzymes such as peroxidases.
Enzyme based microbiosensors enable the determination of non-electroactive compounds. Glucose microbiosensors have been used to measure glucose consumption of Streptococcus mutans biofilms60, and measure the dynamic concentration of endogenous glucose in mammalian brain cells61. Here, we adopted an electrodeposition method to immobilize GOX on a platinum ultramicroelectrode, and the outmost polyurethane coatings effectively protected the immobilized GOX from adsorbing cellulolytic enzymes during degradation experiments, resulting in an improved stability.
LPMOs have been known to act synergistically with glycoside hydrolases during the deconstruction of cellulosic biomass, and supplemented into cellulolytic cocktails in industrial settings of biofuel. The synergy between LPMOs and endoglucanases has been verified using various substrates in different conditions, however, the effect of LPMOs on cellobiohydrolases (exoglucanases) has been disputed by the significant impeding effect of C1-oxidizing LPMOs on reducing-end cellobiohydrolase in recent studies33,34. A mechanistic study found that C1-oxidized cellulose chain ends are poor attack sites for reducing-end cellobiohydrolase (Cel7A) but are preferred by nonreducing-end cellobiohydrolase (Cel6A)34. Our results showed that both TrCel7A and TrCel6A had a higher activity on the wood cell walls oxidized by a C4-oxidizing LPMO9C. Unlike purified cellulosic substrates, the modification of the wood cell wall microstructure is more complex. Since LPMO9C can act on both cellulose chains and hemicellulose networks the increased cellobiohydrolase activity could not only arise from additional ends in the cellulose chain, but also the degradation of the hemicellulose network can increase the accessibility of cellulose chains. This supports that the structural complexity of cell walls affect cellobiohydrolase activity. We found a stronger synergistic activity for the combination of LPMO9C and TrCel7A, which might originate from the formed C4-oxidized cellulose chain ends that are preferred by this cellobiohydrolase starting from the reducing end. Although a systematic investigation of the effect of C4-oxidizing LPMOs on reducing-end cellobiohydrolase activity has not been reported, a promoting effect of C1-C4 oxidizing LPMOs on reducing-end acting Cel7A has been observed33. The effect of LPMO9C in combination with TrCel6A was less pronounced, but agrees with previous studies reporting LPMOs promoting also nonreducing-end cellobiohydrolase activity12,33. For this study on a natural substrate, we have to consider the effect of hemicellulose degradation by LPMO to be beneficial for both cellobiohydrolases. The increased activity of cellobiohydrolases on LPMO-oxidized wood cell walls adds evidence to the claim that LPMOs act in concert with hydrolytic cellulases in the depolymerization of plant biomass.
Methods
Chemicals and instruments
All chemicals were at least of analytical grade and used without further purification. Aqueous solutions were prepared with deionized water (resistivity ca. 18 MΩ cm−1 at 25 °C). The surface microstructure of the polished Pt ultramicroelectrodes and Prussian blue modified Pt ultramicroelectrodes were examined using a scanning electron microscope (FEI Apreo). For the electrochemical experiments, a two-electrode set-up was used with a custom-made miniaturized Ag|AgCl (3 M KCl) as both reference electrode and counter electrode. A micro(bio)sensor served as the working electrode. The miniaturized Ag|AgCl electrode was prepared according to the protocol in our previous study, and was benchmarked to a standard Ag|AgCl reference electrode (Bioanalytical Systems, Inc.). In all the amperometric measurements, data was collected at a rate of 2 s−1 and corrected for background currents of the used micro(bio)sensors.
The SECM system is comprised of a micromanipulator controller (Sutter Instrument) with a piezoelectric positioning module, a potentiostat (Gamry Reference 600+), a lock-in amplifier, and the software (Sensolytics GmbH) to control all parts. The micromanipulator is placed on a stainless-steel board containing a central sample holder inside a custom-made jacketed double Faraday cage, which is mounted on a concrete-filled steel table supported by four active pneumatic dampers (Supplementary Fig. 1a). A digital single-lens reflex camera is connected with the inverted microscope and coupled to the corresponding software for viewing microstructures of the poplar wood slices.
Enzymes
LPMO9C and CDHIIA from N. crassa were recombinantly produced in Pichia pastoris X33 as previously described and purified by a two-step chromatographic procedure employing hydrophobic interaction chromatography (PHE-Sepharose FF) and anion exchange chromatography (DEAE Sepharose FF)62,63. The engineering, production and purification of ChCDH were described in a previous study37. β-glucosidase from Aspergillus niger (Sigma 49291) and glucose oxidase from A. niger (Sigma G7141) were used as purchased. The cellobiohydrolases, β-glucosidase and GOX were dosed on a weight basis. TrCel6A and TrCel7A were purified from the commercial cellulase cocktail (Sigma C2730) following a published procedure. In brief, the solution was desalted using a Sephadex G25-fine column and cellobiohydrolases were purified by anion exchange chromatography (DEAE Sepharose FF). The TrCel7A containing fraction was further purified by size exclusion chromatography (Sephacryl S-200 HR). The TrCel6A containing fraction was subjected to hydrophobic interaction chromatography (PHE Sepharose FF) and finally purified by size exclusion chromatography (Sephacryl S-200 HR). Both cellobiohydrolases were stored in sodium acetate (10 mM, pH 4.8) until further use.
Poplar wood slices
Poplar wood samples were obtained from white poplar (Populus alba) harvested in the winter season (December–February in Vienna, Austria. Small blocks (radial ~5 mm, tangential ~10 mm) were cut out from the branches, debarked and used to prepare 25-µm-thick transverse slices by using a sliding microtome (Microm) with stainless steel blades (Micros). The poplar wood slices were soaked in deionized water for at least 12 h prior to use.
Pt ultramicroelectrodes
Pt ultramicroelectrodes were prepared using a CO2-laser puller (P-2000, Sutter Instrument) following a previously published procedure64. A Pt wire (Ø: 25 µm; Goodfellow) was threaded through and laid at the center of a quartz glass capillary (Øout: 1.0 mm, Øin: 0.5 mm, L: 100 mm), which was fixed in the V-groove of the puller bar in the laser puller. The Pt wire was sealed in quartz capillary by heating (parameters: Heat: 800; Filament: 5; Velocity: 128; Delay: 130; Pull: 0) for 6 cycles (20 s laser on and 40 s laser off). Right afterwards the final pulling step was carried out using typical parameters: (Heat: 510; Filament: 2; Velocity: 85; Delay: 128; Pull: 100). The pulled capillaries with an inlaid Pt wire that crosses the whole microneedle tip were obtained. The unpulled backside of Pt wire was soldered to a copper wire using short soldering tin pieces. A custom-made polishing machine that rotates sandpaper and the electrode individually was used to polish the Pt ultramicroelectrodes’ tip obtaining a disk geometry. The active electrode area was subsequently characterized in 5 mM [Ru(NH3)6]Cl3 containing 100 mM KCl solution. The electrodes with a well-defined amperometric response (plateau in diffusion limited potential region) were used for further fabrication of micro(bio)sensors.
H2O2 microsensor
Prussian blue films were deposited on Pt ultramicroelectrodes by cyclic voltammetry running 10–12 scans from 0.4 to 0.75 V vs. Ag|AgCl with a scan rate of 20 mV s−1 in a solution containing 4 mM FeCl3, 4 mM K3[Fe(CN)6], 0.1 M KCl and 0.1 M HCl. Afterwards, the Prussian blue modified electrodes were rinsed with distilled water and activated by another 20 cycles between −0.05 and +0.35 V in 0.1 M HCl solution with 0.1 M KCl at a scan rate of 50 mV s−1. Their amperometric response at an applied potential of 0.0 V vs. Ag|AgCl was examined in 50 mM sodium acetate buffer solution, pH 5.5 containing varying concentration of H2O2. The Prussian blue modified Pt ultramicroelectrodes with a sensitivity above 60 pA mM−1 were qualified for studying enzymatic degradation of wood cell walls and they were also termed as H2O2 microsensors in this article.
Glucose microbiosensor
Glucose microbiosensors were prepared by immobilization of GOX on the Pt ultramicroelectrodes. GOX was encapsulated within a chitosan polymer matrix by a pH shift-induced deposition of chitosan films65,66. In short, chitosan was dissolved in 0.1 M acetic acid and clarified by centrifugation to remove undissolved solid residues. The final chitosan solution with a concentration of ca. 5 mg mL−1 was adjusted to pH 4.0 by titration with 1 M NaOH solution. A Pt ultramicroelectrode was thread into a Pt wire coil counter electrode in a solution containing 2.5 mg mL−1 chitosan, 15 mg mL−1 GOX and 0.5% glutaraldehyde. A cathodic voltage of −2.5 V was applied using a DC power supply for 60–80 s to precipitate chitosan at the ultramicroelectrode surface and encapsulate enzymes within chitosan films simultaneously. The GOX-modified electrodes were further covered by polyurethane coatings according to a published procedure67.
Constant-distance electrochemical measurements
Positioning of a micro(bio)sensor in close vicinity to wood cell walls was performed by means of shear-force distance control. Two piezo ceramic plates (Piezomechanik, Gmbh) were mounted in close vicinity to the tip end of the quartz glass tube of a micro(bio)sensor through two brass holders (Supplementary Fig. 1d), at an angle of ca. 45° and 1.0–1.5 cm distance from each other. The micro(bio)sensor was mounted to the motorized micromanipulator controller (Fig. 2f). The top piezoelectric plate, driven by an arbitrary waveform generator, induced a low-amplitude vibration, and the bottom plate detected the oscillation amplitude of the micro(bio)sensor tip through a lock-in amplifier that operated in a frequency range of 200–500 kHz. The characteristic resonance frequency of a micro(bio)sensor was determined by comparison of the frequency spectra in air and within the buffer solution (Fig. 2b). A line-scan approach curve was performed until a stop criterion of 5% change in the shear force magnitude was reached (Fig. 2c). The micro(bio)sensor was subsequently retracted to a distance of 25 ± 0.2 μm above the poplar wood slice for real-time measurement of H2O2 or glucose.
The circle sample holder with a glass bottom was used to fix a poplar wood slice (Ø: ~5 mm), which is compressed by the inlaid Teflon ring (Ø: ~3 mm) (Supplementary Fig. 1e). The Teflon ring also serve as an electrochemical cell for all measurements during the enzymatic degradation of wood cell walls. All measurements were conducted in 80 µL of either 50 mM sodium acetate buffer at pH 5.5, or 50 mM sodium phosphate buffer at pH 6.0, containing 50 mM KCl. The humidity inside the Faraday cage was kept at 70–90% to reduce electrolyte evaporation (Supplementary Fig. 1a). Five µL of deionized water was added after every hour to keep the total volume constant.
H2O2-driven LPMO9C oxidation of wood slices
Nine poplar wood slices (thickness: ∼25 µm, diameter: ∼6 mm) were incubated with 100 µM ascorbic acid and 0.5 µM NcLPMO9C in 1.5 ml potassium phosphate buffer (50 mM, pH 6.0). The mixture was kept in a circular shaking motion at 20 rpm at room temperature for at least 10 hours. Aqueous solutions of H2O2 were prepared from 30% hydrogen peroxide at the appropriate concentrations to give a molar feed rate of 10 µmol per hour. At the end, the wood slices were taken out and soaked in 1 M NaCl solution for one hour to release all the bound enzymes. Slices were rinsed thoroughly with deionized water prior to experiments.
Enzyme labeling
Purified CDHIIA and LPMO9C from N. crassa were labeled with DyLight D550 and DyLight D633 N-hydroxy-succinimide (NHS) ester reagents, respectively (Thermo Fisher Scientific). The NHS ester reagents target any exposed α-amine of the N-terminus as well as ε-amines of lysine residues. 2.5 mg of the enzymes were dissolved in 1 mL of 100 mM sodium phosphate-buffered saline, pH 7.4. The enzyme solutions were mixed with DyLight reagents and vortexed to ensure complete mixing. The tubes were covered with aluminum foil and incubated at room temperature (22 °C) for 1 h. The unlabeled dye in the reaction mixture was separated on 250 µL of resin. The molar ratio of DyLight to protein for NcCDHIIA was found to be 7.2 moles of dye per mole of protein and 5.72 moles of dye per mole of NcLPMO9C. The purified and labeled NcCDHIIA and NcLPMO9C were aliquoted and stored at −20 °C in a light-proof environment until further use.
Confocal laser scanning microscope set-up
The transverse poplar wood section was incubated with 1 µM of NcCDHIIA and NcLPMO9C. First, 5 µl of NcLPMO were applied to the section and 3 min later, 5 µL of NcCDHIIA. The enzyme mixture was incubated in the dark and at room temperature for 20 min. Then, the wood section was carefully washed with DI water to remove the excess of unbound enzymes and mounted on a glass slide. A coverslip was placed on top of the specimen and sealed with nail polish to avoid water evaporation. Sections were observed on an inverted confocal laser scanning microscope SP8 (Leica, Germany) with a ×100 1.4 NA oil immersion objective and a zoom factor of 2. The pixel size was set to 0.027 µm with a scan speed of 400 Hz.To minimize possible interference between channels, the images were taken in sequential mode. First, NcLPMO9C labeled with DyLight D633 was imaged with the excitation wavelength of 636 nm and laser intensity of 10 and 100% gain. The emission was collected by a HyD detector with no gaining between 660 and 700 nm. NcCDHIIA labeled with DyLight D550 was observed using an excitation wavelength of 516 nm with a laser intensity of 10%, and the emitted signal was collected by a HyD detector over the range at 560–600 nm with 100% gain. Poplar lignin autofluorescence was excited at 405 nm with a laser intensity of 10%, and the emission was collected by a HyD detector between 415 and 550 nm with 100% gain. In all channels, a line average of 8 was used without frame accumulation. All confocal images were further deconvolved with Huygens Professional v.19.04 (Scientific Volume Imaging). The point spread function was theoretically calculated by LAS X software (Leica, Germany), as well as the background for all channels. The deconvolution was performed with the CMLE algorithm and the signal-to-noise ratio was set to 7 for LPMO and CDH channels and 10 for the lignin channel. The number of iterations was set to 40 with a stop criterion of 0.05.
Data availability
Source data are available for Figs. 1–6 and Supplementary Figs. 2–8. Source data are provided with this paper.
References
Eriksson, K. & Pettersson, B. Oxidation: an important enzyme reaction in fungal degradation of cellulose. FEBS Lett. 49, 282–285 (1974).
Vaaje-Kolstad, G. et al. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 (2010).
Quinlan, R. J. et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl Acad. Sci. USA 108, 15079–15084 (2011).
Harris, P. V. et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 (2010).
Langston, J. A. et al. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77, 7007–7015 (2011).
Agger, J. W. et al. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl Acad. Sci. USA 111, 6287–6292 (2014).
Lo Leggio, L. et al. Structure and boosting activity of a starch-degrading lytic polysaccharide monooxygenase. Nat. Commun. 6, 5961 (2015).
Couturier, M. et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 14, 306–310 (2018).
Hangasky, J. A. & Marletta, M. A. A random-sequential kinetic mechanism for polysaccharide monooxygenases. Biochemistry 57, 3191–3199 (2018).
Hemsworth, G. R., Johnston, E. M., Davies, G. J. & Walton, P. H. Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol. 33, 747–761 (2015).
Beeson, W. T., Vu, V. V., Span, E. A., Phillips, C. M. & Marletta, M. A. Cellulose degradation by polysaccharide monooxygenases. Annu. Rev. Biochem. 84, 923–946 (2015).
Keller, M. B. et al. Promoting and impeding effects of lytic polysaccharide monooxygenases on glycoside hydrolase activity. ACS Sustain. Chem. Eng. 8, 14117–14126 (2020).
Bissaro, B. et al. Molecular mechanism of the chitinolytic peroxygenase reaction. Proc. Natl Acad. Sci. USA 117, 1504–1513 (2020).
Jagadeeswaran, G., Veale, L. & Mort, A. J. Do lytic polysaccharide monooxygenases aid in plant pathogenesis and herbivory? Trends Plant Sci. 26, 142–155 (2021).
Kracher, D. et al. Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352, 1098–1101 (2016).
Phillips, C. M., Beeson, W. T., Cate, J. H. & Marletta, M. A. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 (2011).
Tan, T. C. et al. Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat. Commun. 6, 7542 (2015).
Beeson, W. T., Phillips, C. M., Cate, J. H. D. & Marletta, M. A. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 (2012).
Chylenski, P. et al. Lytic polysaccharide monooxygenases in enzymatic processing of lignocellulosic biomass. ACS Catal. 9, 4970–4991 (2019).
Bissaro, B. et al. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 13, 1123–1128 (2017).
Müller, G., Chylenski, P., Bissaro, B., Eijsink, V. G. H. & Horn, S. J. The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol. Biofuels 11, 1–18 (2018).
Hangasky, J. A., Iavarone, A. T. & Marletta, M. A. Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl Acad. Sci. USA 115, 4915–4920 (2018).
Jones, S. M., Transue, W. J., Meier, K. K., Kelemen, B. & Solomon, E. I. Kinetic analysis of amino acid radicals formed in H2O2-driven CuI LPMO reoxidation implicates dominant homolytic reactivity. Proc. Natl Acad. Sci. USA 117, 2–8 (2020).
Kont, R., Bissaro, B., Eijsink, V. G. H. & Väljamäe, P. Kinetic insights into the peroxygenase activity of cellulose-active lytic polysaccharide monooxygenases (LPMOs). Nat. Commun. 11, 5786 (2020).
Hedison, T. M. et al. Insights into the H2O2-driven catalytic mechanism of fungal lytic polysaccharide monooxygenases. FEBS J. 288, 4115–4128 (2021).
Rovira, C., Walton, P. H., Wang, B., Wang, Z. & Davies, G. J. Activation of O2 and H2O2 by lytic polysaccharide monooxygenases. ACS Catal. 10, 12760–12769 (2020).
Vermaas, J. V., Crowley, M. F., Beckham, G. T. & Payne, C. M. Effects of lytic polysaccharide monooxygenase oxidation on cellulose structure and binding of oxidized cellulose oligomers to cellulases. J. Phys. Chem. B 119, 6129–6143 (2015).
Hofrichter, M. & Ullrich, R. Oxidations catalyzed by fungal peroxygenases. Curr. Opin. Chem. Biol. 19, 116–125 (2014).
Sugano, Y. & Yoshida, T. Dyp-type peroxidases: recent advances and perspectives. Int. J. Mol. Sci. 22, 5556 (2021).
Tian, C. et al. Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl Acad. Sci. USA 106, 22157–22162 (2009).
Eibinger, M. et al. Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 (2014).
Himmel, M. E., Ding, S., Johnson, D. K. & Adney, W. S. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–808 (2007).
Tokin, R., Ipsen, J. Ø., Westh, P. & Johansen, K. S. The synergy between LPMOs and cellulases in enzymatic saccharification of cellulose is both enzyme- and substrate-dependent. Biotechnol. Lett. 42, 1975–1984 (2020).
Keller, M. B. et al. A comparative biochemical investigation of the impeding effect of C1-oxidizing LPMOs on cellobiohydrolases. J. Biol. Chem. 296, 100504 (2021).
Phillips, C. M., Iavarone, A. T. & Marletta, M. A. Quantitative proteomic approach for cellulose degradation by Neurospora crassa. J. Proteome Res. 10, 4177–4185 (2011).
Jun, H., Kieselbach, T. & Jönsson, L. J. Enzyme production by filamentous fungi: analysis of the secretome of Trichoderma reesei grown on unconventional carbon source. Microb. Cell Fact. 10, 1–10 (2011).
Kracher, D. et al. Polysaccharide oxidation by lytic polysaccharide monooxygenase is enhanced by engineered cellobiose dehydrogenase. FEBS J. 287, 897–908 (2020).
Salazar, P., Martín, M., O’Neill, R. D., Roche, R. & González-Mora, J. L. Improvement and characterization of surfactant-modified Prussian blue screen-printed carbon electrodes for selective H2O2 detection at low applied potentials. J. Electroanal. Chem. 674, 48–56 (2012).
Clausmeyer, J., Actis, P., López Córdoba, A., Korchev, Y. & Schuhmann, W. Nanosensors for the detection of hydrogen peroxide. Electrochem. commun. 40, 28–30 (2014).
Karyakin, A. A., Gitelmacher, O. V. & Karyakina, E. E. Prussian Blue-based first-generation biosensor. A sensitive amperometric electrode for glucose. Anal. Chem. 67, 2419–2423 (1995).
Sygmund, C. et al. Semi-rational engineering of cellobiose dehydrogenase for improved hydrogen peroxide production. Microb. Cell Fact. 12, 1–10 (2013).
Filandr, F. et al. The H2O2-dependent activity of a fungal lytic polysaccharide monooxygenase investigated with a turbidimetric assay. Biotechnol. Biofuels 13, 1–13 (2020).
Haddad Momeni, M. et al. Discovery of fungal oligosaccharide-oxidising flavo-enzymes with previously unknown substrates, redox-activity profiles and interplay with LPMOs. Nat. Commun. 12, 2132 (2021).
Bissaro, B., Várnai, A., Røhr, Å. K. & Eijsink, V. G. H. Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass. Microbiol. Mol. Biol. Rev. 82, 1–51 (2018).
Kadić, A., Várnai, A., Eijsink, V. G. H., Horn, S. J. & Lidén, G. In situ measurements of oxidation–reduction potential and hydrogen peroxide concentration as tools for revealing LPMO inactivation during enzymatic saccharification of cellulose. Biotechnol. Biofuels 14, 1–10 (2021).
Gusakov, A. V., Bulakhov, A. G., Demin, I. N. & Sinitsyn, A. P. Monitoring of reactions catalyzed by lytic polysaccharide monooxygenases using highly-sensitive fluorimetric assay of the oxygen consumption rate. Carbohydr. Res. 452, 156–161 (2017).
Kracher, D., Andlar, M., Furtmüller, P. G. & Ludwig, R. Active-site copper reduction promotes substrate binding of fungal lytic polysaccharide monooxygenase and reduces stability. J. Biol. Chem. 293, 1676–1687 (2018).
Takahashi, Y. et al. Topographical and electrochemical nanoscale imaging of living cells using voltage-switching mode scanning electrochemical microscopy. Proc. Natl Acad. Sci. USA 109, 11540–11545 (2012).
Koley, D., Ramsey, M. M., Bard, A. J. & Whiteley, M. Discovery of a biofilm electrocline using real-time 3D metabolite analysis. Proc. Natl Acad. Sci. USA 108, 19996–20001 (2011).
Eijsink, V. G. H. et al. On the functional characterization of lytic polysaccharide monooxygenases (LPMOs). Biotechnol. Biofuels 12, 1–16 (2019).
Daniel, G. et al. Characteristics of Gloeophyllum trabeum alcohol oxidase, an extracellular source of H2O2 in brown rot decay of wood. Appl. Environ. Microbiol. 73, 6241–6253 (2007).
Guillén, F., Martínez, A. T., Martínez, M. J. & Evans, C. S. Hydrogen-peroxide-producing system of Pleurotus eryngii involving the extracellular enzyme aryl-alcohol oxidase. Appl. Microbiol. Biotechnol. 41, 465–470 (1994).
Bouws, H., Wattenberg, A. & Zorn, H. Fungal secretomes - nature’s toolbox for white biotechnology. Appl. Microbiol. Biotechnol. 80, 381–388 (2008).
Kracher, D. et al. Inter-domain electron transfer in cellobiose dehydrogenase: Modulation by pH and divalent cations. FEBS J. 282, 3136–3148 (2015).
Breslmayr, E. et al. Inhibition of the peroxygenase lytic polysaccharide monooxygenase by carboxylic acids and amino acids. Antioxidants 11, 1096 (2022).
Giardina, P., Palmieri, G., Fontanella, B., Rivieccio, V. & Sannia, G. Manganese peroxidase isoenzymes produced by Pleurotus ostreatus grown on wood sawdust. Arch. Biochem. Biophys. 376, 171–179 (2000).
Ambert-Balay, K., Dougherty, M. & Tien, M. Reactivity of manganese peroxidase: site-directed mutagenesis of residues in proximity to the porphyrin ring. Arch. Biochem. Biophys. 382, 89–94 (2000).
Rieder, L., Stepnov, A. A., Sørlie, M. & Eijsink, V. G. H. Fast and specific peroxygenase reactions catalyzed by fungal mono-copper enzymes. Biochemistry 60, 3633–3643 (2021).
Kuusk, S. et al. Kinetics of H2O2-driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase. J. Biol. Chem. 293, 523–531 (2018).
Jayathilake, N. M. & Koley, D. Glucose microsensor with covalently immobilized glucose oxidase for probing bacterial glucose uptake by scanning electrochemical microscopy. Anal. Chem. 92, 3589–3597 (2020).
Smith, S. K. et al. Simultaneous voltammetric measurements of glucose and dopamine demonstrate the coupling of glucose availability with increased metabolic demand in the rat striatum. ACS Chem. Neurosci. 8, 272–280 (2017).
Kittl, R., Kracher, D., Burgstaller, D., Haltrich, D. & Ludwig, R. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79 (2012).
Sygmund, C. et al. Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 78, 6161–6171 (2012).
Botz, A. et al. Local activities of hydroxide and water determine the operation of silver-based oxygen depolarized cathodes. Angew. Chem. Int. Ed. 57, 12285–12289 (2018).
Kurzawa, C., Hengstenberg, A. & Schuhmann, W. Immobilization method for the preparation of biosensors based on pH shift-induced deposition of biomolecule-containing polymer films. Anal. Chem. 74, 355–361 (2002).
Smith, S. K. et al. Quantitative comparison of enzyme immobilization strategies for glucose biosensing in real-time using fast-scan cyclic voltammetry coupled with carbon-fiber microelectrodes. ChemPhysChem 19, 1197–1204 (2018).
Fang, L. et al. A needle-type glucose biosensor based on PANI nanofibers and PU/E-PU membrane for long-term invasive continuous monitoring. Biosens. Bioelectron. 97, 196–202 (2017).
Acknowledgements
We thank Wolfgang Schuhmann for valuable discussions and support in setting up the SECM and Monika Debreczeny for help with the confocal laser scanning microscope. We thank Christopher Schulz for providing the material and protocol for the polyurethane coating. This work was supported by European Research Council through European Union’s Horizon 2020 research and innovation program (ERC Consolidator Grant OXIDISE) under grant agreement no. 726396.
Author information
Authors and Affiliations
Contributions
H.C. and R.L. designed the study. H.C., N.G.A., A.B., and F.C. performed the experiments. H.C., D.K., F.C., L.S., N.G.A., S.S., and R.L. interpreted data. H.C. and R.L. wrote the initial manuscript. All authors contributed in revising and writing the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kiyohiko Igarashi, Edson Nossol, and Jiwei Zhang for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, H., Gacias Amengual, N., Botz, A. et al. Investigating lytic polysaccharide monooxygenase-assisted wood cell wall degradation with microsensors. Nat Commun 13, 6258 (2022). https://doi.org/10.1038/s41467-022-33963-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33963-w
This article is cited by
-
Assessing the role of redox partners in TthLPMO9G and its mutants: focus on H2O2 production and interaction with cellulose
Biotechnology for Biofuels and Bioproducts (2024)
-
Enhancing enzymatic saccharification yields of cellulose at high solid loadings by combining different LPMO activities
Biotechnology for Biofuels and Bioproducts (2024)
-
Molecular-level carbon traits underlie the multidimensional fine root economics space
Nature Plants (2024)
-
Mutational dissection of a hole hopping route in a lytic polysaccharide monooxygenase (LPMO)
Nature Communications (2024)
-
Initial characterization of an iron superoxide dismutase from Thermobifida fusca
JBIC Journal of Biological Inorganic Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
