
Abstract
Enterotoxigenic E. coli (ETEC) produce heat-labile (LT) and/or heat-stable (ST) enterotoxins, and commonly cause diarrhea in resource-poor regions. ETEC have been linked repeatedly to sequelae in children including enteropathy, malnutrition, and growth impairment. Although cellular actions of ETEC enterotoxins leading to diarrhea are well-established, their contributions to sequelae remain unclear. LT increases cellular cAMP to activate protein kinase A (PKA) that phosphorylates ion channels driving intestinal export of salt and water resulting in diarrhea. As PKA also modulates transcription of many genes, we interrogated transcriptional profiles of LT-treated intestinal epithelia. Here we show that LT significantly alters intestinal epithelial gene expression directing biogenesis of the brush border, the major site for nutrient absorption, suppresses transcription factors HNF4 and SMAD4 critical to enterocyte differentiation, and profoundly disrupts microvillus architecture and essential nutrient transport. In addition, ETEC-challenged neonatal mice exhibit substantial brush border derangement that is prevented by maternal vaccination with LT. Finally, mice repeatedly challenged with toxigenic ETEC exhibit impaired growth recapitulating the multiplicative impact of recurring ETEC infections in children. These findings highlight impacts of ETEC enterotoxins beyond acute diarrheal illness and may inform approaches to prevent major sequelae of these common infections including malnutrition that impact millions of children.
Similar content being viewed by others


Introduction
Infectious diarrhea remains a leading cause of death and morbidity among young children in low-middle-income countries where access to clean water and sanitation remains in short supply1. Enterotoxigenic E. coli (ETEC), initially discovered as a cause of severe, cholera-like illness2, are one of the most common pathogens associated with moderate-severe diarrhea among children under the age of 5 years3,4, and are perennially the most common cause of diarrhea in travelers5 to endemic regions where these organisms are thought to account for hundreds of millions of cases of diarrheal illness each year6.
Importantly, ETEC infections have been linked to non-diarrheal sequelae, including “environmental enteric dysfunction (EED),” a condition characterized by impaired nutrient absorption, impaired growth7,8, and malnutrition9,10, adding significantly to the morbidity as well as deaths from diarrhea and other infections11. The risk of stunting multiplies with each episode of diarrheal illness in children under the age of two years12, a period during which children residing in impoverished areas commonly sustain multiple ETEC infections8. However, the molecular pathogenesis underlying the intestinal changes associated with EED, and the contribution of individual pathogens, including ETEC, remain poorly understood.
Similarly, toxin-producing E. coli have also been repeatedly identified in patients with tropical sprue13,14,15, a condition classically described in adults residing for extended periods of time in areas where ETEC diarrheal disease is common. Like EED, tropical sprue is associated with changes to the small intestinal villous architecture, including ultrastructural alteration of the epithelial brush border formed by the microvilli16,17, nutrient malabsorption, and wasting.
The basic molecular mechanisms underpinning acute watery diarrhea caused by ETEC are well-established18. ETEC produces heat-labile (LT) and/or heat-stable (ST) enterotoxins that activate the production of cAMP and cGMP second messengers, respectively, leading to activation of cellular kinases that in turn modulate the activity of sodium and chloride channels in the apical membrane of intestinal epithelial cells to promote net efflux of salt and water into the intestinal lumen resulting in watery diarrhea.
LT and cholera toxin (CT) share ~85% amino acid identity, and both toxins exert their major effects on the cell through the ADP-ribosylation of the alpha subunit of Gs (Gsα), a stimulatory intracellular guanine nucleotide-binding protein. Inhibition of Gsα GTPase activity leads to constitutive activation of adenylate cyclase and increased production of intracellular cAMP19.
In its central role as a second messenger, cAMP governs a diverse array of cellular processes20 and modulates the transcription of multiple genes through a number of cAMP-responsive transcriptional activators and repressors21. cAMP activates protein kinase A (PKA), a heterotetramer, by liberating its two regulatory subunits from the catalytic subunits, which are then free to phosphorylate a wide variety of cytoplasmic and nuclear protein substrates22. PKA largely regulates transcription by phosphorylation of transcription factors, including the cyclic AMP response element binding protein (CREB) and the cAMP-response element modulator (CREM), which bind cAMP-response elements (CRE) in the promoter regions of target genes21,22,23.
Notably, cholera toxin (CT), LT, and dibutyryl-cyclic AMP all induce hypersecretion and impact the architecture of gastrointestinal epithelia in rodent small intestine24, while small intestinal biopsies of patients with acute cholera exhibit marked changes in the ultrastructure of the intestinal brush border, the major absorptive surface in the small intestine25,26, including shortening and disruption of the microvilli. Consistent with these observations, studies of young children less than two years of age in Bangladesh have specifically associated LT-producing ETEC with undernutrition27, suggesting that heat-labile toxin may exert effects on intestinal mucosa that extend beyond acute diarrheal illness.
Here we demonstrate that in addition to the canonical effects of LT on the cellular export of salt and water into the intestinal lumen, this toxin impacts multiple genes involved in the formation of microvilli, resulting in marked alteration of the architecture of the intestinal brush border, the major site of nutrient absorption in the small intestine. These effects are compounded by the alteration of solute transporters within the brush border epithelia, potentially disrupting the absorption of multiple essential nutrients.
Results
Heat-labile toxin markedly alters the transcriptomes ofintestinal epithelial cells
Commensurate with the importance of cAMP as a second messenger, we found that compared to either untreated controls or cells treated with a catalytically inactive (E211K) mutant of LT, wild-type LT holotoxin substantially modulated transcription of many genes in intestinal epithelial cells. In RNA-seq studies of polarized Caco-2 intestinal epithelial cells, we found that 3832 genes were significantly (p ≤ 10−5) upregulated and 3687 downregulated in response to LT, while the inactive toxin failed to induce significant changes in the transcriptome (Fig. 1a). However, Caco-2 cells are derived from distant metastases of a colon cancer tumor in which transcriptomes would likely be altered relative to untransformed intestinal epithelia28,29, and cAMP signaling is known to be aberrant in some transformed cells23. Therefore, to examine a more physiologically relevant target, we next examined the impact of LT on differentiated small intestinal enteroids. Here, we found that far fewer genes were differentially expressed (≤10−5), with 746 significantly upregulated, and 561 downregulated in response to intoxication with LT (Fig. 1b). Notably, however, we found substantial statistically significant overlap in genes significantly modulated in Caco-2 cells and enteroids (Supplementary Table 3 and Supplementary Dataset 1) with the transcription of hundreds of genes significantly up- or down-regulated in both groups. Gene ontology enrichment analysis, as well as ontology-independent investigation of genes (CompBio, Supplementary Fig. 1) modulated by the toxin, highlighted multiple cellular components associated with both the development and function of the absorptive surface of the small intestine (Supplementary Dataset 2).

Model at left depicts the E. coli heat-labile toxin92 based on PDB structure 1LTS with the A1 subunit in blue, the A2 region in yellow, and pentameric B subunit in green. The E211K mutation of mLT is in the active site of the A1 subunit. a Scatterplot of RNAseq data right depicts differential expression profiles of Caco-2 cells following exposure to a heat-labile toxin (n = 2) relative to untreated cells (n = 2) and cells treated with the biologically inactive mLT (n = 2). (Because expression profiles of untreated and mLT-treated cells were virtually identical, their combined expression profiles totaling n = 4 replicates are compared here to LT-treated cells). b RNA-seq data from polarized small intestinal ileal enteroids treated with LT (n = 3) compared to control untreated (n = 3) cells. Differentially expressed genes were identified by DESeq293.
Heat-labile toxin impairs the development of small intestinal microvilli
Small intestinal enterocytes are each covered with hundreds of microvilli, complex structures comprised of a central core of actin filaments within protrusions of the plasma membrane. Collectively, the luminal surface of the intestine formed by these microvilli, known as the brush border, represents the major absorptive surface of the gastrointestinal tract. Three major classes of proteins are required for the biogenesis of microvilli30 (Fig. 2a). These include (1) proteins such as villin, epsin, plastin, and EPS8 that bundle parallel clusters of actin filaments; BAIP2L1 (IRTKS) responsible for recruiting the EPS bundling protein to the tips of microvilli31; (2) ezrin, myo1a32, myo6 that link the actin cytoskeleton with the plasma membrane; and (3) protocadherin molecules CDHR2 and CDHR5 engaged in extracellular heterotypic complexes between the tips of the microvilli30 that are stabilized by a tripartite complex of MYO7B, ANKS4B33, and USH1C33. Interrogation of transcriptional profiles indicated that the transcription of each of these classes of genes was significantly altered following exposure to wild-type heat-labile toxin (Fig. 2b). Similarly, RT-PCR confirmed decreased expression of multiple genes involved in microvilli biogenesis, including VIL1 encoding villin (Fig. 3a), and we were able to demonstrate that production of villin was depressed in polarized small intestinal enteroids (Fig. 3b and Supplementary Fig. 2). In addition, TEM images of polarized small intestinal enteroids exposed to heat-labile toxin demonstrated significantly shortened and disorganized microvillus structures on the apical surface of enterocytes (Fig. 3c).

a Diagram at the top (adapted from ref. 30) depicts molecules involved in key elements of microvillus development. b Heatmaps of RNA-seq data obtained following treatment of Caco-2 intestinal cells (left) with mLT (n = 2 biologically independent samples) or LT (n = 2) relative to untreated cells (n = 2); and ileal enteroids (right) treated with LT (n = 3) relative to control untreated cells (n = 3). Comparisons were made with DESeq293. Bars indicate absolute log2 fold change values + SE. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 10−4, and *****p ≤ 10−5.

a Quantitative RT-PCR data from n = 9 biologically independent samples for selected microvillus genes comparing untreated (−) and LT-treated (+) ileal epithelial cells (enteroid line 235D). b Villin production is suppressed in small intestinal enteroids following treatment (t + 18 h) with LT (100 µg/ml). Shown are representative confocal images obtained showing membrane (CellMask, blue), nuclei (white), villin (gold), and merged image. The graph at right shows apical villin geometric mean fluorescence intensity data relative to the corresponding cytoplasmic signal. Each symbol (n = 12) represents a unique region of interest. For a, b ****p < 0.0001, ***p < 0.001 by Mann–Whitney two-tailed testing. c TEM images of small intestinal microvilli following treatment LT (right) compared to control untreated cells (left). The graph at right shows the length of microvilli when enteroids (n = 2 biologically independent samples) are treated before (pre) and after (post) differentiation on polarized ileal cells ****<0.0001 by ANOVA (Kruskal–Wallis, nonparametric testing).
Heat-labile toxin modulates the transcription of multiple brush border nutrient transport genes
The human solute carrier (SLC) gene superfamily is comprised of more than 50 gene families thought to encode more than 300 functional transporters34. Many of the SLC proteins are enriched in the small intestinal brush border, where they transport critical nutrients, including amino acids, oligopeptides, sugars, and vitamins. We found that transcription of many SLC genes was altered in Caco-2 cells as well as small intestinal enteroids (Fig. 4a). These included transporters for Zinc, known both to be deficient in children with enteropathy35, and a micronutrient critical for intestinal homeostasis. Likewise, transcription of SLC19A3 encoding the principal SLC responsible for uptake of the water-soluble B vitamin thiamine (vitamin B1)36 by differentiated intestinal epithelial cells lining the surface of intestinal villi of the proximal small intestine37,38 (Fig. 4b) was repressed as was the production of the corresponding protein (supplementary figure 3a). Moreover, we found that LT treatment of human small intestinal organoids also interfered with the transcription of the cis-regulatory element specificity protein 1 (SP1) previously shown to govern the transcription of SLC19A339,40,41 (Fig. 4c and Supplementary Fig. 3b). Finally, we found that thiamine transport was significantly depressed following exposure to LT (Fig. 4d) providing additional evidence that ETEC can impair transport of critical nutrients.

a Heatmap indicating key SLC genes modulated by heat-labile toxin (LT) compared to enzymatically inactive E112K LT mutant (mLT), or untreated (ø) Caco-2 cells (left) and human small intestinal (ileal) enteroids (Hu235D, right). Bars indicate absolute log2 fold change values + SE. Comparisons were made with DESeq293. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 10−4, *****p ≤ 10−5. Real-time qRT-PCR confirming LT-mediated modulation of genes in ileal (Hu235D) enteroids encoding b the major thiamine transporter SLC19A3 and c the SP1 cis-regulatory element. Data reflect two independent experiments with two replicates each. d Uptake of [3H]-thiamine by Hu235D cells is impaired following LT treatment. Data presented in b–d are from two independent experiments with n = 3 replicates each. (*<0.05, **<0.01 by Mann–Whitney two-tailed comparisons).
LT-producing ETEC disrupts the absorptive architecture of the small intestine
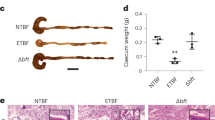
To further study the potential impact of ETEC toxins on intestinal architecture, we performed challenge studies in infant mice. Compared to sham-challenged (PBS) controls, or mice challenged with a toxin-deficient strain of ETEC, we again noted down-regulation of genes involved in F-actin bundling, membrane cross-linking, and intermicrovillus adhesion complex formation, all required for intestinal microvilli (Fig. 5a) biogenesis. Likewise, on examination of small intestinal villi we found that the production of villin in enterocyte brush borders was substantially decreased relative to sham-challenged controls (Fig. 5b, c). In mice challenged with a wild-type ETEC isolate that makes ST and LT, but not an LT/ST-toxin-negative mutant (jf4763, Supplementary Table 1), we observed significant alteration in the architecture of the intestinal brush border with significant shortening and disorganization of the microvilli (Fig. 5d, e and Supplementary Fig. 4a–d) reminiscent of the earlier ultrastructural studies of patients with tropical sprue17 and V. cholerae infections25. Similarly, we found that in mice challenged with a strain containing an isogenic mutation in eltA (jf571, Supplementary Table 1) encoding the LT A subunit, which still makes heat-stable toxins, the microvillus architecture was preserved (Fig. 5f, g), suggesting that LT is the principal toxin underlying the enteropathic changes to the enterocyte surfaces.

Timeline at the top depicts the challenge with ETEC or control nontoxigenic isolate or sham (PBS) challenge. a Quantitative PCR results for genes involved in brush border development in small intestinal samples obtained from infant mice (n = 9/group) 7 days after challenge with toxigenic ETEC (jf876), nontoxigenic ETEC (jf4763, LT−/ST−) PBS controls. Comparisons between data represent ANOVA, Kruskal–Wallis testing where ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. b Immunofluorescence images of small intestinal sections showing villin expression (blue), and nuclei (white). c Mean villin fluorescence intensity normalized per enterocyte (n = 4 mice/group) ****p ≤ 0.0001 by Mann–Whitney (two-tailed) nonparametric comparisons. d Representative transmission electron microscopy (TEM) images of the small intestinal brush border from mice challenged with toxin-negative (∆, left) and toxigenic ETEC (wt, right). e Microvillus length ****p ≤ 0.0001 by Mann–Whitney (two-tailed) nonparametric comparisons. Data represent geometric mean length from n = 5 mice per group in three independent experiments. f TEM images from mice challenged with jf570 (eltA::KmR), sham PBS controls, or mice challenged with wild-type ETEC. g Length of microvilli (dashed horizontal lines represent geometric means). ****p < 0.0001 by Kruskal–Wallis.
Despite the dramatic toxin-dependent changes to the absorptive surface of the intestine, the early growth kinetics of suckling mice challenged a single time with either wild-type bacteria or a heat-labile toxin deletion mutant were surprisingly similar (Supplementary Fig. 5A) and paralleled those of sham-challenged controls. However, enteropathy in young children is thought to reflect damage elicited by repeated infections. Children in endemic regions typically suffer multiple ETEC infections before their second birthday, and the risk of enteropathic sequelae increases multiplicatively per episode8,12. Therefore, to assess the contribution of repeated ETEC infections to growth impairment, we compared the growth kinetics of suckling mice challenged a single time to those repeatedly infected with wild-type toxigenic ETEC. These studies demonstrated a clear impact of repeated infection on growth (Supplementary Fig. 5B). Finally, we found that the growth kinetics of mice repeatedly challenged with wild-type ETEC H10407 was significantly retarded relative to those challenged with the isogenic LT-mutant jf876 (Supplementary Fig. 5C). Therefore, repeated infections in this model appear to recapitulate impacts observed following repeated ETEC infection in children, and our data suggest that these features are at least in part driven by LT.
Maternal vaccination with LT prevents brush border disruption
To address whether enteropathic changes to the small intestine can be prevented by vaccination, and to further define the role of LT, we vaccinated mouse dams with heat-labile toxin and examined the brush border ultrastructure in suckling mice. Vaccinated dams but not sham vaccinated controls expressed significant levels of IgA and IgG in breast milk (Fig. 6a), consistent with increased levels of antibodies in the stomachs of infant mice (Fig. 6b). Notably, we found that maternal vaccination with LT completely abrogated changes to the microvilli (Fig. 6c, d) further substantiating the importance of LT in driving changes to the epithelial architecture.

Timeline depicts vaccination and challenge (top): maternal intranasal (i.n.) vaccinations with 10 µg LT/immunization (yellow arrows on days 0, 14, 28) and neonatal challenge at 3 days of age (green arrow) followed by sacrifice and tissue collection at 7 days post-infection (gray arrow). a Kinetic ELISA data of from triplicate samples of breast milk anti-LT (IgA, and IgG in n = 2 immunized dams and 1 un-immunized control). *<0.05 by Mann–Whitney two-tailed nonparametric testing. b Anti-LT antibodies in the gastric contents of neonatal mice at day 53. ****p < 0.0001 by Mann–Whitney two-tailed comparisons. c Representative transmission electron microscopy images of brush border microvilli from unvaccinated mice challenged with wild-type ETEC (left), vaccinated mice challenged with wild-type ETEC, and vaccinated un-challenged controls. d Microvillus lengths (based on image analysis of n = 5 mice group) ****<0.0001 Kruskal–Wallis comparisons.
LT modulates key transcription factors that govern enterocyte development
Despite the marked alteration in transcription mediated by LT, the majority of genes critical for brush border development lacked conserved CRE sites23. Therefore, we performed transcription factor target enrichment analysis42,43 to identify potential transcription factors responsible for differential regulation of genes significantly (P < 0.05) upregulated or downregulated by LT in both Caco-2 and enteroid RNA-seq datasets (Supplementary Table 4 and Supplementary Dataset 1). The upregulated genes were most significantly (p = 4.2 × −10−3) linked to transcription factor targets of AP-1 encoded by the c-jun gene, previously shown to be regulated by cAMP44, and to be involved in intestinal epithelial repair45. Notably, however, downregulated genes were most significantly enriched in targets for the HNF4α transcription factor or its intestine-specific paralog HNF4γ (P = 1.9 × −10−4) that were recently shown to regulate multiple genes required for brush border development46,47. Importantly, PKA has also been shown to phosphorylate HNF4 α at a consensus recognition site within the DNA binding domain, shared with HNF4 γ, inhibiting transcription48.
Of note, the transcription of both paralogs was found to be significantly depressed following exposure of intestinal epithelia to LT (Fig. 7a, b), as were levels of HNF4γ in nuclear fractions from toxin-treated cells (Fig. 7c), suggesting that activation of cAMP can interfere with transcription mediated by HNF4. To further examine the impact of LT on HNF4-mediated transcription, we introduced a transcriptional reporter plasmid containing six tandem copies of the HNF4 transcriptional response element (5′-CAAAGGTCA-3′) linked to a human codon-optimized Gaussia princeps luciferase into Caco-2 cells. These assays demonstrated that HNF4-mediated transcription was dramatically reduced in cells treated with LT (Fig. 7d). Similarly, we found that relative to nuclei of intestinal epithelial cells in ileal segments from sham-challenged control mice, those from ETEC-challenged mice exhibited significantly less HNFγ (Fig. 7e) further supporting a role for ETEC in modulating the production of this important transcription factor. Chen et al described a “feed-forward regulatory module” essential to enterocyte differentiation in which HNF4 and SMAD4 transcription factors reciprocally activate each other’s transcription. As would be predicted from this model, we found that transcription of SMAD4 was also impaired by LT (Supplementary Fig. 6A, B), leading us to speculate that LT-mediated phosphorylation of HNF4 by PKA, interrupts this critical transcription module (Supplementary Fig. 6C–E).

a Heatmap demonstrating the impact of LT on transcription of paralogous transcription factors HNF4α and HNF4γ in Caco-2 cells (left) and ileal enteroids, (Hu235D, right) ø untreated, mLT mutant LT. b qRT-PCR (TaqMan) data confirming decreased transcription of HNF4 transcription factors following treatment of enteroids with LT (n = 9 biologically independent samples). ****<0.0001 by Mann–Whitney two-tailed comparisons. c HNF4γ is decreased in nuclear fractions obtained from small intestinal enteroids following treatment with LT. Shown in the HNF4γ immunoblot are samples from four independent experiments, with the graph below-representing quantitation of signal intensity normalized to the lamin B1 nuclear protein (p = 0.0017, paired t-test, one-tailed). Bars indicate mean ± 95% confidence intervals. d HNF4 transcription Gaussia luciferase reporter assay showing a decrease in signal following treatment of TR104-transfected Caco-2 cells with LT. 2 experimental replicates (n = 15 samples total) ***p < 0.001 Wilcoxon matched pairs, one-tailed). e Confocal microscopy of representative Ileal sections from sham-challenged (PBS) left, and ETEC-infected mice (right). Immunofluorescence intensity of HNF4γ signal in sections from n = 5 control mice, and n = 6 ETEC-challenged mice. Membranes (yellow) were strained with CellMask orange (Thermo Fisher C10045), nuclei (blue) were stained with DAPI, and HNFγ immunostaining was represented in white. Each symbol represents a microscopic region of interest. Bars represent geographic means (p < 0.0001, Mann–Whitney two-tailed comparisons).
Collectively, the current studies demonstrate that in addition to their known canonical effects on ion transport that culminate in watery diarrhea, ETEC toxins can drive appreciable derangement of enterocyte architecture and function by interfering with key pathways in intestinal epithelia that govern the formation of mature enterocytes capable of effective nutrient absorption. These findings have important implications for our understanding and prevention of enteropathic conditions linked to ETEC.
Discussion
Understanding the molecular events that lead to sequelae of undernutrition and growth faltering following ETEC infections may be key to the effective design of prevention strategies, including vaccines49. Although the molecular mechanisms involved in the fluid and ion fluxes into the intestinal lumen leading to diarrhea are firmly established, only recently has evidence emerged to suggest that ETEC toxins may incite previously unappreciated changes in small intestinal epithelia50,51. The present studies, initiated to identify additional effects of LT, were prompted by an appreciation that cyclic nucleotides, particularly cAMP, govern a multitude of cellular pathways, potentially resulting in collateral impacts that extend beyond the acute episodes of diarrhea. Consistent with this model, we found that exposure of intestinal cells to heat-labile toxin altered the transcription of hundreds of genes. A central theme highlighted in the analysis of these transcriptional alterations is that LT affects major classes of genes involved in the biogenesis of microvilli and the function of the intestinal brush border, the major site of nutrient uptake in the small intestine, potentially offering a direct molecular link to sequelae of malnutrition and impaired growth in children.
The potential clinical relevance of the observations reported here is highlighted by remarkably similar ultrastructural alteration of intestinal epithelial cells seen in small intestinal biopsies of patients with acute cholera25 and tropical sprue17. In both entities, the brush border was noted to be abnormal, with shortened, irregular microvilli. Importantly, however, despite the structural and functional similarity between LT and CT, clinical cholera, unlike ETEC infections, has not been linked to enteropathy or attendant sequelae. Whether this relates to the repetitive nature of ETEC infections compared to the durable protective immunity that follows a single V. cholerae infection52 is not presently clear. Similarly, while tropical sprue remains a leading cause of malabsorption in regions where infectious diarrhea is prevalent53,54,55, the most debilitating forms of this illness have not typically followed isolated cases of traveler’s diarrhea, but occur in resident populations or expatriates56 repeatedly assailed by diarrhea while residing in endemic regions57. Likewise, our data also highlight the potential importance of repeated infections on the development of sequelae.
The negative impacts of LT on brush border architecture, with a commensurate reduction in surface area available for nutrient absorption, are compounded by the alteration of multiple SLC genes that encode transporters critical for the uptake of essential vitamins and other molecules. Importantly, small intestinal biopsies obtained from Zambian children with enteropathy and refractory stunting exhibited similar changes in SLC gene expression profiles58. The decreased transcription of molecules required for intestinal zinc uptake, in cells treated with LT is particularly intriguing given the known aberrations in zinc absorption in children with enteropathy35,59,60, the possible contribution of zinc deficiency to enteropathic changes61, and the salutary effects of zinc in the treatment of children with diarrhea62,63.
Further study will be needed to precisely delineate the role of ETEC LT and ST enterotoxins, alone and in combination, in driving enteropathic changes to the intestine and sequelae. ST-producing ETEC were most strongly associated with moderate to severe diarrhea in GEMS3, and follow-on studies of children enrolled in these studies have also linked infections with ETEC encoding heat-stable toxin to growth faltering7. However, our earlier analysis of a global collection of more than 1100 ETEC isolates, including those collected in GEMS, demonstrated that slightly more than half of all ST-encoding strains also encoded LT, and that roughly one-third of the isolates overall encoded LT alone, ST-LT, or ST only64. While LT-producing ETEC have been specifically linked to malnutrition among children in Bangladesh27, and our in vitro and animal studies point to the potential importance of LT, additional effort will be needed to correlate toxin-induced morphologic and functional perturbation of the intestinal brush border with outcomes in children. Importantly, the long-term morbidity associated with ETEC infections does not appear to correlate with the severity of diarrhea as both mild illness4, and perhaps asymptomatic colonization may lead to growth faltering.
Further refinement of animal models that can faithfully recapitulate features of enteropathy are also needed. Indeed, conventional mice lack genes that could be required to reproduce the full effects of ETEC. For instance, each of the carcinoembryonic antigen cell adhesion molecules (CEACAMs) that are substantially upregulated on human small intestinal epithelia in response to LT, and which we have recently shown to play a critical role in ETEC interactions with human small intestine50, are absent in mice.
While the precise mechanism underlying LT-mediated modulation of genes required for microvillus biogenesis and absorptive function of the brush border is presently unclear, stimulation of adenylate cyclase invokes many cAMP-responsive nuclear factors that may serve either to activate or repress transcription. Genes implicated in the development of microvilli are mostly devoid of consensus palindromic (TGACGTCA) or “half” (CGTCA) cAMP-response element (CRE) sites within their promoter regions for direct modulation by CREB65, which typically is involved as a transcriptional activator, and both CREB and CREM can yield several alternatively spliced variants that may act as either activators or repressors66. cAMP second messaging also engages multiple signaling pathways converging at CREB67, and PKA can phosphorylate and modulate the activity of multiple transcription factors to act either as transcriptional activators or repressors, including SP168. Notably, putative binding sites for HNF4, a cAMP-modulated transcription factor48 known to regulate genes needed for the formation of microvilli46,47, were significantly enriched in the promotors of genes downregulated by LT. Both HNF4α and HNF4γ possess canonical PKA recognition sites within their DNA binding motifs, and PKA phosphorylation of these sites interrupts transcription69. HNF4 activates the transcription of SMAD4, and in turn, SMAD4 activates the transcription of HNF470. Both transcription factors then engage genes needed for the effective differentiation of stem cells to mature enterocytes70. Modulation of the activity of this transcription factor by LT would therefore be predicted to have a marked impact on pathways critical to intestinal epithelial homeostasis.
We should also note that increases in cellular cAMP can impact multiple cellular pathways independent of PKA. Included among these are pathways governed by a more recently discovered family of cellular cAMP-binding molecules, exchange proteins activated by cAMP (EPACs). EPACs appear to play critical roles as guanine exchange factors that regulate GTPase proteins71 and are involved in complex signaling networks implicated in cell growth, differentiation, and morphogenesis72,73.
cAMP can also exert potent epigenetic influences on transcription. CREB-binding protein (CBP) possesses intrinsic histone acetyl-transferase (HAT) activity74, and can therefore modulate chromatin remodeling, enhancing access to transcription factors. In addition, cAMP messaging through PKA leads to phosphorylation-dependent activation of the histone demethylase enzyme PHF2 to promote the transcription of multiple genes that can impact the transition from stem cells to epithelial cells75.
Increased intestinal permeability is a recognized hallmark of enteropathy in young children in LMICs76. Given the known negative impacts of cholera toxin on epithelial barrier function77, and its structural and functional similarity to a heat-labile toxin, LT may exert additional enteropathic effects beyond the impact on brush border biogenesis described here.
Altogether it seems likely that multiple pathways governed by increases in cellular cAMP may underlie the morphologic and functional disruption of the brush border epithelial observed in our studies. Nevertheless, the data presented here provide compelling evidence that the heat-labile toxin of ETEC ultimately impacts multiple genes required for the biogenesis and function of the brush border, the major site of nutrient absorption in the human small intestine. The findings may have significant implications for our understanding of sequelae linked to ETEC, including environmental enteropathy in young children, and tropical sprue in adults. The increased acknowledgement of long-term morbidity linked to ETEC and an improved understanding of the role of ETEC enterotoxins as drivers of this morbidity may also strengthen the case for vaccines49 specifically engineered to prevent both acute illness and sequelae.
Methods
The studies reported here comply with all relevant ethical regulations and the study protocols have been approved by the IACUC, Institutional Biosafety, and Institutional Review Boards at Washington University in Saint Louis School of Medicine.
Culture and differentiation of enteroids
Archived small intestinal enteroid specimens derived from biopsy samples of adult patients undergoing routine endoscopy were obtained from the Organoids Core of the Digestive Disease Research Core Center (DDRCC) at Washington University School of Medicine under Institutional Review Board (IRB) protocol number 201406083. Cell lines used in these are routinely validated for response to the heat-labile toxin by cAMP assays (Arbor Assays, Ann Arbor, MI).
Purified crypt cells from the ileum (Hu235D) were resuspended in Matrigel (BD Biosciences, San Jose, CA, USA) and 15 µL of resuspended matrix gel was added to each well in 24 well plates. Plates were incubated at 37 °C and 5% CO2 with 50% L-WRN conditioned media (CM) and 50% primary culture medium (Advanced DMEM/F12, Invitrogen) supplemented with 20% FBS, 2 mM l-glutamine, 100 units/mL penicillin, 0.1 mg/mL streptomycin, 10 µM Y-27632 (ROCK inhibitor, Tocris Bioscience, R&D systems, Minneapolis, MN, USA), and 10 µM SB431541 (TGFBR1 inhibitor, Tocris Bioscience, R&D systems).
To induce differentiation and polarization of enteroids, cells were washed once to remove Matrigel, followed by trypsinization and centrifugation at 1100 × g for 5 min. Cells were then resuspended in 1:1 CM and primary medium with Y-27632 and SB431541 as described above. Cells were then plated on semiporous filters (Transwells®, 6.5 mm insert, 24 well plate, 0.4-μm polyester membrane, Corning Incorporated, Kennebunk, ME, USA) that had been previously coated with Collagen IV (Millipore Sigma). Transwell® inserts were rinsed with DMEM/F12 with Hepes, 10% FBS, l-glutamine, Penicillin, and streptomycin. Cells were allowed to grow to confluency in 50% conditioned media (CM) and then changed to a differentiation medium (5% CM in primary medium + ROCK inhibitor) for 48 h. Differentiated cells were used for toxin treatment.
Propagation of Caco-2 cells
Caco-2 intestinal epithelial cells were obtained from ATCC (ATCC HTB-37) and cultured at 37 °C, 5% CO2, in Eagle’s MEM supplemented with 20% of fetal bovine serum (FBS). To generate polarized monolayers, ~1 × 105 cells were seeded onto polystyrene membrane filters (0.4 μM, 6.5 mm diameter insert, Transwell, Corning) and grew for at least a week prior to toxin treatment. Media was replaced with fresh media every two days.
Toxin treatment
Polarized differentiated cells were treated with a heat-labile toxin (LT) in a differentiation medium (100 ng/mL) in a volume of 700 µL at the basolateral side of the Transwell insert and 100 µL volume on the apical aspect of the monolayer. Treated cells were incubated at 37 °C for 18 h and then fixed for transmission electron microscopy or fluorescence microscopy, or lysed for RNA extraction (GE Healthcare, Buckinghamshire, UK). LT and mutant LT (E211K, mLT) were kindly provided by Dr. John D. Clements, Tulane University, New Orleans, Louisianna, USA.
RNA extraction and cDNA synthesis
RNA was extracted using the Illustra RNAspin Mini RNA extraction kit (GE Healthcare, Buckinghamshire, UK). Three biological replicates per treatment (untreated and LT treated) were submitted to the Genome Technology Access Center (GTAC) at Washington University in St. Louis School of Medicine for RNA-seq library preparation. An Agilent Bioanalyzer was used to determine the integrity of RNA samples. cDNA was generated using SuperscriptTM ViloTM cDNA synthesis kit (Invitrogen by Thermo Fisher) after normalizing RNA concentrations.
RNA-seq library preparation
Library preparation was performed with 10 ng of total RNA, integrity was determined using an Agilent bioanalyzer. ds-cDNA was prepared using the SMARTer Ultra Low RNA kit for Illumina Sequencing (Clontech) per the manufacturer’s protocol. cDNA was fragmented using a Covaris E220 sonicator using peak incident power 18, duty factor 20%, cycles/burst 50, and time 120 s. cDNA was blunt-ended, had an A base added to the 3′ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12–15 cycles using primers incorporating unique index tags. Fragments were sequenced on an Illumina HiSeq-2500 using single reads extending 50 bases.
RNA-seq data processing and analysis
RNA-seq reads were aligned to the Ensembl top-level human genome assembly with STAR version 2.7.3a. Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.54.1. Read counts were used as input for DESeq2 differential gene expression analysis (version 1.24.0)78 with default settings, and a minimum P value significance threshold of 10−5 (after false discovery rate [FDR79] correction for the number of tests). Fragments per kilobase per million reads mapped (FPKM) values for relative gene expression were calculated from DESeq2-normalized read counts, and Z-scores were calculated per gene using the average and standard deviations of FPKM values across samples. Log2 fold changes were identified from differential expression output. Pathway enrichment analysis for KEGG80 and Gene Ontology (GO)81 pathways among gene sets of interest was performed using the over-representation analysis tool provided on the WebGestalt42 web server (version 2019). Heatmap and bar graph visualization was performed with Microsoft Excel.
Transcript counts were produced by Sailfish version 0.6.3. Sequencing performance was assessed for the total number of aligned reads, the total number of uniquely aligned reads, genes and transcripts detected, ribosomal fraction known junction saturation, and read distribution over known gene models with RSeQC version 2.3.
All gene-level and transcript counts were then imported into the R/Bioconductor package EdgeR and TMM normalization size factors were calculated to adjust for samples for differences in library size. Ribosomal features, as well as any feature not expressed in at least the smallest condition size minus one sample were excluded from further analysis and TMM size factors were recalculated to created effective TMM size factors. The TMM size factors and the matrix of counts were then imported into R/Bioconductor package Limma and weighted likelihoods based on the observed mean-variance relationship of every gene/transcript and sample were then calculated for all samples with the voomWithQualityWeights function. The performance of the samples was assessed with a spearman correlation matrix and multidimensional scaling plots. Gene/transcript performance was assessed with plots of the residual standard deviation of every gene to their average log count with a robustly fitted trend line of the residuals. Generalized linear models were then created to test for gene/transcript level differential expression. Differentially expressed genes and transcripts were then filtered for FDR-adjusted p values less than or equal to 0.05.
The biological interpretation of the large set of features found in the Limma results were then elucidated for global transcriptomic changes in known Gene Ontology (GO) and KEGG terms with the R/Bioconductor packages GAGE and Pathview. Briefly, GAGE measures for perturbations in GO or KEGG terms based on changes in observed log2 fold changes for the genes within that term versus the background log2 fold changes observed across features not contained in the respective term as reported by Limma. For GO terms with an adjusted statistical significance of FDR ≤0.05, heatmaps were automatically generated for each respective term to show how genes co-vary or co-express across the term in relation to a given biological process or molecular function. In the case of KEGG-curated signaling and metabolism pathways, Pathview was used to generate annotated pathway maps of any perturbed pathway with an unadjusted statistical significance of p value ≤0.05. Genes significantly modulated in both Caco-2 cells and enteroids were subjected to further ontology-free analysis via CompBio v1.4 (PercayAI, Inc., www.percayai.com/compbio)82 to identify unifying biological themes in sets of genes differentially expressed between pairwise comparisons of different groups.
Quantitative real-time PCR
Quantitative real-time PCR was performed using a QuantStudio 3 real-time detection system (Applied Biosystems). Fast SYBR Green master mix (Applied Biosystems/Thermo Fisher) was used for qPCR analysis. Disassociation curve analysis was performed to assess the specificity of amplification for each sample, and PCR product size was verified by agarose gel electrophoresis. Percent expression was normalized by GAPDH and analyzed using the comparative threshold cycle (Ct) method. Amplification of SLC19A3 transcripts from small intestinal enteroids was performed as recently described83 with relative gene expression normalized to β actin. HNF4a, HNFg, and SMAD4 gene expression was determined by TaqMan (Thermo Fisher) using validated primer and probe sets. Primers and TaqMan probes used in this study are listed in Supplemental Table 2.
Transcriptional reporter assays
To assess the impact of LT on HNF4-mediated transcription, pTR104 (GeneCopoeia) carrying six copies of the HNF4 transcriptional response element upstream of a secreted Gaussia luciferase (Gluc) reporter gene was transfected (FuGENE, Promega) into confluent Caco-2 cells. Media (EMEM, ATCC) were exchanged 24 h after transfection, and after an additional 24 h, baseline samples of media were removed from each well and stored at −80 °C. Wells were then treated with a heat-labile toxin (100 ng/ml) overnight (16 h). At the end of treatment cell culture supernatants media were transferred to black-walled microplates (Greiner 655086) and processed as directed (Secrete-Pair Luminescence, GeneCopoeia, Inc. Rockville, MD), followed by luminescence detection (Synergy H1, BioTek). Data were expressed as Relative Light Units (RLU) before and after treatment with LT.
SMAD4-mediated transcription was assessed by transient transfection of Caco-2 cells with the SBE4-luc plasmid containing four copies of the SMAD4 transcriptional response element 5’-GTCTAGAC-3'84.
Following treatment with overnight treatment with LT, cells were resuspended in 80 µl of EMEM media and mixed with an equal volume of ONEGLO EX Reagent (Nano-Glo® Dual-Luciferase® Reporter Assay System, Promega) for 20 min on an orbital shaker at 300 rpm, then read on the luminometer as above.
Immunoblotting
SLC19A3, SP1
Total protein was extracted from LT toxin-treated (100 ng/ml) human differentiated enteroid monolayers (235D line) and untreated control using radioimmunoprecipitation assay (RIPA) buffer (Sigma) containing protease inhibitor cocktail. An equal amount (∼25 μg) of the proteins were loaded on a NuPAGE 4-12% Bris–Tris gradient gels (Invitrogen) as previously described83, then blotted onto polyvinylidene difluoride (PVDF) membranes and probed with anti-SLC19A3 (1:1000; Cat# 13407-1-AP; Proteintech), or anti-SP1 (1:1000; Cat# ab124804; Abcam) antibodies and together with anti-beta actin (1:3000; Cat# sc-47778) primary antibodies. The specificity of the SLC19A3 antibodies was validated in our laboratory previously using different approaches that include overexpression of tagged protein or gene silencing85. Anti-SP1 antibodies were validated by the manufacturer using knockout cell lysate protein samples. The SLC19A3 and SP1 protein bands from the blot were then identified with corresponding anti-rabbit IR-800 dye (1:30,000; Cat# 926-32211; LI-COR Bioscience) and anti-mouse IR-680 dye (1:30,000; Cat# 926-68020; LI-COR Bioscience) secondary antibodies incubation for 1 h at room temperature. Relative expression of specific proteins was calculated by comparing the fluorescence intensities in an Odyssey infrared imaging system (LI-COR Bioscience) with respect to the corresponding beta-actin signal.
Villin
Differentiated LT-treated and control enteroid monolayers were lysed using NE-PERTM nuclear and cytoplasmic extraction reagents (Thermo Scientific). Cell membrane pellets were solubilized in PBS containing 1% Triton X-100 supplemented with a protease inhibitor cocktail (PierceTM protease inhibitor mini, Thermo Scientific). Equal amounts of total protein were loaded on a 4–20% gradient SDS-PAGE gel (Mini Protean TGX, Bio-Rad), then blotted onto nitrocellulose membranes and probed with anti-villin mouse monoclonal antibody (1:1000; Cat# SC-66022; Santa Cruz Biotechnology) followed by detection with HRP-conjugated anti-mouse secondary antibody (1:1000; Cat# 7076 S; Cell Signaling Technology). Blots were developed with Clarity Western ECL substrate (Bio-Rad) and imaged with a c600 imaging system (Azure Biosystems). Relative Villin expression signals were then analyzed with respect to corresponding Coomassie-stained gel using ImageJ analysis software.
Thiamin uptake
Initial rates (30 min; at 37 °C) of carrier-mediated thiamin uptake were examined in LT-treated (100 ng/ml; overnight) and untreated control differentiated small intestinal enteroid monolayers incubated in Krebs–Ringer buffer (pH 7.4) containing [3H]-thiamin (15 nM). Enteroid monolayers were then washed with ice-cold Krebs–Ringer buffer, followed by lysis with NaOH and neutralization with 10 N HCl. The radioactive content was counted using a liquid scintillation counter as described previously83. Uptake of thiamin by its respective and distinct carrier-mediated mechanism was determined by subtracting uptake of [3H]-thiamin in the presence of a high pharmacological concentration (1 mM) of unlabeled thiamin from uptake in their absence; all uptake data points were calculated relative to total protein content (in milligrams) of the different preparations and presented as a percentage relative to simultaneously performed controls.
Confocal microscopy
Cell monolayers were fixed with 2% paraformaldehyde for 30 min at 37 °C and then washed 3× with PBS prior to blocking with 1% BSA in PBS for 1 h at room temperature. Villin expression was detected using 1:100 dilution of anti-villin antibodies raised in mice (Santa Cruz Biotechnology) for 1 h at room temperature, followed by additional 1 h incubation with 1:200 dilution of fluorescence-tagged goat anti-mouse IgG Alexa Fluor 488 (Thermo Fisher) (Supplemental Table 5). Cell membranes were stained with 1:2000 dilution of CellMask orange (Invitrogen) and nuclei were counterstained with DAPI (1:1000) and mounted on glass slides using Prolong Gold antifade reagent (Invitrogen). Images were captured and analyzed on a Nikon C2 confocal microscope equipped with NIS-Elements AR 5.11.01 software (Nikon).
Electron microscopy
For ultrastructural analyses, in vitro-grown differentiated polarized monolayers of human ileal enteroid samples, as well as mouse intestinal biopsy samples, were fixed in 2% paraformaldehyde/2.5% glutaraldehyde (Ted Pella, Inc., Redding, CA) in 100 mM sodium cacodylate buffer, pH 7.2 for 2 h at room temperature and then overnight at 4 °C. Samples were washed in sodium cacodylate buffer and postfixed in 2% osmium tetroxide (Ted Pella, Inc) 1 h at room temperature. Samples were then rinsed in dH20, dehydrated in a graded series of ethanol, and embedded in Eponate 12 resin (Ted Pella, Inc.). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc., Peabody, MA) equipped with an AMT 8 megapixel digital camera and AMT Image Capture Engine V602 software (Advanced Microscopy Techniques, Woburn, MA). Images were analyzed using ImageJ software for microvilli length and structures.
Identification of potential transcription factor target sites
The CREB targets database65 (http://natural.salk.edu/CREB/) was used to identify potential categorical CRE full (TGACGTCA) or half (TGACG/CGTCA) sites within the promotor regions of genes modulated by the heat-labile toxin. Following previously described protocol23, 5 kilobase upstream sequences from each human gene were retrieved from Ensembl86 and were searched for the CREB-binding sites TGACGTCA and TGACGTAA (and their reverse complements), based on sequences retrieved from the TRANSFAC87 database (matrix ID: V$CREB_01). CREB sequence matches were identified in the upstream sequences of 1,302 genes. WebGestalt42 was used for overall functional enrichment of potential transcription factor binding sequences upstream from genes that were significantly (p < 0.05) up- and down-regulated in both Caco-2 cells and enteroids.
Suckling mouse challenge studies
Studies in mice were performed under protocol 20-0438 approved by the Washington University in Saint Louis Institutional Animal Care and Use Committee. In neonatal mouse challenge studies, 3-day-old CD-1 mice (male and female) were inoculated with wild-type ETEC (H10407), isogenic toxin deletion mutants, or sterile PBS (sham negative controls). Inocula were prepared from frozen glycerol stocks maintained at −80 °C grown overnight (~16 h) in 2 ml Luria Bertani (LB) broth in a shaking incubator (at 37 °C, 225 rpm). Cultures were then diluted 1:100 in 20 ml of fresh LB, grown for an additional 2 h, and bacteria were harvested by centrifugation (6000 rpm, 5 min at 4 °C). Pellets were then washed once in ice-cold sterile PBS, then resuspended to a density of ~1 × 105 colony forming units (CFU) per 20 µl inoculum. CFU in each inoculum were determined by plating serial dilutions LB-agar plates. Mice were then inoculated with ETEC directly into the stomach through the abdominal wall using a 31 g insulin needle. Following infection, mice were marked with tattoo ink to define groups and returned to the cage. Seven days post-infection, mice were sacrificed, and the ileal tissue collected was fixed for microscopy or preserved for RNA extraction.
Bacterial strain construction
The strains used in these studies are described in Supplementary Table 1. Strain jf4763 is devoid of all enterotoxins and was constructed from a previously engineered ST-negative mutant of H10407, jf284888. Briefly, jf2848 was first transformed with pKD46, a lambda red recombinase helper plasmid with selection on ampicillin. Jf2848(pKD46) was then transformed with a PCR amplicon generated with primers _to replace the eltAB genes encoding LT with a spectinomycin resistance cassette resulting in jf4763. The strain was subsequently validated by PCR and by testing supernatants in GM1 ganglioside ELISA to confirm the lack of LT production.
Vaccination and immunologic assessment
Adult female mouse dams were vaccinated intranasally three times at 2-week intervals with 10 µg LT holotoxin in PBS, with the last dose administered ~2 weeks prior to anticipated delivery. Control mice were mock vaccinated with PBS. Fecal pellets collected from vaccinated dams were used to assess mucosal responses to LT. To collect breast milk, the dam was separated from the litter ~2 h prior to milking. Oxytocin (Sigma O3251), 0.002 IU/g, was injected IP to stimulate milk production, followed by collection via pipette tip. Anti-LT immune responses in maternal fecal extracts and breast milk, and stomach contents and sera of neonates (male and female) were assessed by GM1 ganglioside89 kinetic ELISA90 for anti-LT IgA and IgG, using plates coated with GM1 (Sigma G7641), as previously described91.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNAseq data were deposited at the Sequence Read Archive (SRA) on the NCBI website https://www.ncbi.nlm.nih.gov/sra under BioProject accession number PRJNA875141. Primary source data and original images are available on Figshare as detailed in Supplementary Table 6 of the supplementary information.
References
Collaborators, G. B. D. D. D. Quantifying risks and interventions that have affected the burden of diarrhoea among children younger than 5 years: an analysis of the Global Burden of Disease Study 2017. Lancet Infect. Dis. 20, 37–59 (2020).
Sack, R. B. et al. Enterotoxigenic Escherichia coli isolated from patients with severe cholera-like disease. J. Infect. Dis. 123, 378–385 (1971).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222 (2013).
Kotloff, K. L. et al. The incidence, aetiology, and adverse clinical consequences of less severe diarrhoeal episodes among infants and children residing in low-income and middle-income countries: a 12-month case-control study as a follow-on to the Global Enteric Multicenter Study (GEMS). Lancet Glob. Health 7, e568–e584 (2019).
Steffen, R., Hill, D. R. & DuPont, H. L. Traveler’s diarrhea: a clinical review. JAMA 313, 71–80 (2015).
Khalil, I. A. et al. Morbidity and mortality due to shigella and enterotoxigenic Escherichia coli diarrhoea: the Global Burden of Disease Study 1990-2016. Lancet Infect. Dis. 18, 1229–1240 (2018).
Nasrin, D. et al. Pathogens associated with linear growth faltering in children with diarrhea and impact of antibiotic treatment: the Global Enteric Multicenter Study. J. Infect. Dis. https://doi.org/10.1093/infdis/jiab434 (2021).
Qadri, F. et al. Disease burden due to enterotoxigenic Escherichia coli in the first 2 years of life in an urban community in Bangladesh. Infect. Immun. 75, 3961–3968 (2007).
Black, R. E., Brown, K. H. & Becker, S. Effects of diarrhea associated with specific enteropathogens on the growth of children in rural Bangladesh. Pediatrics 73, 799–805 (1984).
Anderson, J. D. T. et al. Burden of enterotoxigenic Escherichia coli and shigella non-fatal diarrhoeal infections in 79 low-income and lower middle-income countries: a modelling analysis. Lancet Glob. Health 7, e321–e330 (2019).
Caulfield, L. E., de Onis, M., Blossner, M. & Black, R. E. Undernutrition as an underlying cause of child deaths associated with diarrhea, pneumonia, malaria, and measles. Am. J. Clin. Nutr. 80, 193–198 (2004).
Checkley, W. et al. Multi-country analysis of the effects of diarrhoea on childhood stunting. Int J. Epidemiol. 37, 816–830 (2008).
Klipstein, F. A., Short, H. B., Engert, R. F., Jean, L. & Weaver, G. A. Contamination of the small intestine by enterotoxigenic coliform bacteria among the rural population of Haiti. Gastroenterology 70, 1035–1041 (1976).
Klipstein, F. A., Holdeman, L. V., Corcino, J. J. & Moore, W. E. Enterotoxigenic intestinal bacteria in tropical sprue. Ann. Intern. Med. 79, 632–641 (1973).
Klipstein, F. A., Engert, R. F. & Short, H. B. Enterotoxigenicity of colonising coliform bacteria in tropical sprue and blind-loop syndrome. Lancet 2, 342–344 (1978).
Brunser, O., Eidelman, S. & Klipstein, F. A. Intestinal morphology of rural Haitians. A comparison between overt tropical sprue and asymptomatic subjects. Gastroenterology 58, 655–668 (1970).
Mathan, M., Mathan, V. I. & Baker, S. J. An electron-microscopic study of jejunal mucosal morphology in control subjects and in patients with tropical sprue in southern India. Gastroenterology 68, 17–32 (1975).
Fleckenstein, J. M. et al. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 12, 89–98 (2010).
Tsai, S. C., Noda, M., Adamik, R., Moss, J. & Vaughan, M. Enhancement of choleragen ADP-ribosyltransferase activities by guanyl nucleotides and a 19-kDa membrane protein. Proc. Natl Acad. Sci. USA 84, 5139–5142 (1987).
Lodish, H, B. A., Zipursky, S. L., Matsudaira, P., Baltimore, D. &Darnell, J. Molecular Cell Biology (W. H. Freeman, 2000).
Sassone-Corsi, P. Transcription factors responsive to cAMP. Annu Rev. Cell Dev. Biol. 11, 355–377 (1995).
Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a011148 (2012).
Zambon, A. C. et al. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc. Natl Acad. Sci. USA 102, 8561–8566 (2005).
Hansson, H. A., Lange, S., Lonnroth, I., Rozell, B. & Tinberg, H. Cytoskeletal disarrangement in rat intestinal epithelium after in vivo exposure to secretagogues. Acta Pathol. Microbiol. Immunol. Scand. [B] 92, 93–99 (1984).
Mathan, M. M., Chandy, G. & Mathan, V. I. Ultrastructural changes in the upper small intestinal mucosa in patients with cholera. Gastroenterology 109, 422–430 (1995).
Asakura, H., Tsuchiya, M., Watanabe, Y., Enomoto, Y. & Morita, A. Electron microscopic study on the jejunal mucosa in human cholera. Gut 15, 531–544 (1974).
Platts-Mills, J. A. et al. Association between enteropathogens and malnutrition in children aged 6-23 mo in Bangladesh: a case-control study. Am. J. Clin. Nutr. 105, 1132–1138 (2017).
Ilantzis, C., DeMarte, L., Screaton, R. A. & Stanners, C. P. Deregulated expression of the human tumor marker CEA and CEA family member CEACAM6 disrupts tissue architecture and blocks colonocyte differentiation. Neoplasia 4, 151–163 (2002).
Tadjali, M., Seidelin, J. B., Olsen, J. & Troelsen, J. T. Transcriptome changes during intestinal cell differentiation. Biochim. Biophys. Acta 1589, 160–167 (2002).
Crawley, S. W., Mooseker, M. S. & Tyska, M. J. Shaping the intestinal brush border. J. Cell Biol. 207, 441–451 (2014).
Postema, M. M., Grega-Larson, N. E., Neininger, A. C. & Tyska, M. J. IRTKS (BAIAP2L1) elongates epithelial microvilli using EPS8-dependent and independent mechanisms. Curr. Biol. 28, 2876–2888 e2874 (2018).
Tyska, M. J. et al. Myosin-1a is critical for normal brush border structure and composition. Mol. Biol. Cell 16, 2443–2457 (2005).
Crawley, S. W., Weck, M. L., Grega-Larson, N. E., Shifrin, D. A. Jr. & Tyska, M. J. ANKS4B is essential for intermicrovillar adhesion complex formation. Dev. Cell 36, 190–200 (2016).
He, L., Vasiliou, K. & Nebert, D. W. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum. Genomics 3, 195–206 (2009).
Manary, M. J. et al. Perturbed zinc homeostasis in rural 3-5-y-old Malawian children is associated with abnormalities in intestinal permeability attributed to tropical enteropathy. Pediatr. Res. 67, 671–675 (2010).
Reidling, J. C., Lambrecht, N., Kassir, M. & Said, H. M. Impaired intestinal vitamin B1 (thiamin) uptake in thiamin transporter-2-deficient mice. Gastroenterology 138, 1802–1809 (2010).
Subramanian, V. S., Marchant, J. S. & Said, H. M. Targeting and trafficking of the human thiamine transporter-2 in epithelial cells. J. Biol. Chem. 281, 5233–5245 (2006).
Said, H. M., Balamurugan, K., Subramanian, V. S. & Marchant, J. S. Expression and functional contribution of hTHTR-2 in thiamin absorption in human intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G491–498 (2004).
Nabokina, S. M., Subramanian, V. S., Valle, J. E. & Said, H. M. Adaptive regulation of human intestinal thiamine uptake by extracellular substrate level: a role for THTR-2 transcriptional regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G593–599 (2013).
Nabokina, S. M. & Said, H. M. Characterization of the 5′-regulatory region of the human thiamin transporter SLC19A3: in vitro and in vivo studies. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G822–829 (2004).
Nabokina, S. M., Reidling, J. C. & Said, H. M. Differentiation-dependent up-regulation of intestinal thiamin uptake: cellular and molecular mechanisms. J. Biol. Chem. 280, 32676–32682 (2005).
Wang, J., Vasaikar, S., Shi, Z., Greer, M. & Zhang, B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137 (2017).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Nakamura, T., Datta, R., Sherman, M. L. & Kufe, D. Regulation of c-jun gene expression by cAMP in HL-60 myeloid leukemia cells. J. Biol. Chem. 265, 22011–22015 (1990).
Wang, P. Y. et al. c-Jun enhances intestinal epithelial restitution after wounding by increasing phospholipase C-gamma1 transcription. Am. J. Physiol. Cell Physiol. 312, C367–C375 (2017).
San Roman, A. K., Aronson, B. E., Krasinski, S. D., Shivdasani, R. A. & Verzi, M. P. Transcription factors GATA4 and HNF4A control distinct aspects of intestinal homeostasis in conjunction with transcription factor CDX2. J. Biol. Chem. 290, 1850–1860 (2015).
Chen, L. et al. The nuclear receptor HNF4 drives a brush border gene program conserved across murine intestine, kidney, and embryonic yolk sac. Nat. Commun. 12, 2886 (2021).
Dankel, S. N., Hoang, T., Flageng, M. H., Sagen, J. V. & Mellgren, G. cAMP-mediated regulation of HNF-4alpha depends on the level of coactivator PGC-1alpha. Biochim. Biophys. Acta 1803, 1013–1019 (2010).
Khalil, I. et al. Enterotoxigenic Escherichia coli (ETEC) vaccines: priority activities to enable product development, licensure, and global access. Vaccine 39, 4266–4277 (2021).
Sheikh, A. et al. CEACAMs serve as toxin-stimulated receptors for enterotoxigenic Escherichia coli. Proc. Natl Acad. Sci. USA 117, 29055–29062 (2020).
Sheikh, A. et al. Enterotoxigenic Escherichia coli degrades the host MUC2 mucin barrier to facilitate critical pathogen-enterocyte interactions in human small intestine. Infect. Immun. https://doi.org/10.1128/IAI.00572-21 (2021).
Harris, J. B. Cholera: immunity and prospects in vaccine development. J. Infect. Dis. 218, S141–S146 (2018).
Pipaliya, N. et al. Spectrum of chronic small bowel diarrhea with malabsorption in Indian subcontinent: is the trend really changing? Intest. Res. 14, 75–82 (2016).
Ghoshal, U. C., Srivastava, D., Verma, A. & Ghoshal, U. Tropical sprue in 2014: the new face of an old disease. Curr. Gastroenterol. Rep. 16, 391 (2014).
Dutta, A. K., Balekuduru, A. & Chacko, A. Spectrum of malabsorption in India–tropical sprue is still the leader. J. Assoc. Physicians India 59, 420–422 (2011).
Klipstein, F. A. & Falaiye, J. M. Tropical sprue in expatriates from the tropics living in the continental United States. Medicine 48, 475–491 (1969).
Klipstein, F. A. Tropical sprue in travelers and expatriates living abroad. Gastroenterology 80, 590–600 (1981).
Kelly, P. et al. Gene expression profiles compared in environmental and malnutrition enteropathy in Zambian children and adults. EBioMedicine 70, 103509 (2021).
Zyambo, K. et al. Reduced fractional absorption of zinc in children with environmental enteropathy in Zambia. J. Pediatr. Gastroenterol. Nutr. 74, 277–283 (2022).
Long, J. M. et al. Zinc absorption from micronutrient powders is low in Bangladeshi toddlers at risk of environmental enteric dysfunction and may increase dietary zinc requirements. J. Nutr. 149, 98–105 (2019).
Lindenmayer, G. W., Stoltzfus, R. J. & Prendergast, A. J. Interactions between zinc deficiency and environmental enteropathy in developing countries. Adv. Nutr. 5, 1–6 (2014).
Roy, S. K. et al. Impact of zinc supplementation on intestinal permeability in Bangladeshi children with acute diarrhoea and persistent diarrhoea syndrome. J. Pediatr. Gastroenterol. Nutr. 15, 289–296 (1992).
Dhingra, U. et al. Lower-dose zinc for childhood diarrhea - A randomized, multicenter trial. N. Engl. J. Med. 383, 1231–1241 (2020).
Kuhlmann, F. M. et al. Conservation and global distribution of non-canonical antigens in enterotoxigenic Escherichia coli. PLoS Negl. Trop. Dis. 13, e0007825 (2019).
Zhang, X. et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl Acad. Sci. USA 102, 4459–4464 (2005).
Mayr, B. & Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 (2001).
Delghandi, M. P., Johannessen, M. & Moens, U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal 17, 1343–1351 (2005).
Rohlff, C., Ahmad, S., Borellini, F., Lei, J. & Glazer, R. I. Modulation of transcription factor Sp1 by cAMP-dependent protein kinase. J. Biol. Chem. 272, 21137–21141 (1997).
Viollet, B., Kahn, A. & Raymondjean, M. Protein kinase A-dependent phosphorylation modulates DNA-binding activity of hepatocyte nuclear factor 4. Mol. Cell. Biol. 17, 4208–4219 (1997).
Chen, L. et al. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat. Genet. 51, 777–785 (2019).
de Rooij, J. et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477 (1998).
Bos, J. L., de Rooij, J. & Reedquist, K. A. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2, 369–377 (2001).
Frische, E. W. & Zwartkruis, F. J. Rap1, a mercenary among the Ras-like GTPases. Dev. Biol. 340, 1–9 (2010).
Ogryzko, V. V., Schiltz, R. L., Russanova, V., Howard, B. H. & Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87, 953–959 (1996).
Pattabiraman, D. R. et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680 (2016).
Amadi, B. et al. Adaptation of the small intestine to microbial enteropathogens in Zambian children with stunting. Nat. Microbiol. 6, 445–454 (2021).
Guichard, A. et al. Cholera toxin disrupts barrier function by inhibiting exocyst-mediated trafficking of host proteins to intestinal cell junctions. Cell Host Microbe 14, 294–305 (2013).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Gene Ontology, C. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021).
Delannoy-Bruno, O. et al. Evaluating microbiome-directed fibre snacks in gnotobiotic mice and humans. Nature 595, 91–95 (2021).
Sabui, S. et al. Hypoxia inhibits colonic uptake of the microbiota-generated forms of vitamin B1 via HIF-1alpha-mediated transcriptional regulation of their transporters. J. Biol. Chem. 298, 101562 (2022).
Zawel, L. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 (1998).
Anandam, K. Y., Srinivasan, P., Yasujima, T., Al-Juburi, S. & Said, H. M. Proinflammatory cytokines inhibit thiamin uptake by human and mouse pancreatic acinar cells: involvement of transcriptional mechanism(s). Am. J. Physiol. Gastrointest. Liver Physiol. 320, G108–G116 (2021).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Matys, V. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–110 (2006).
Zhu, Y. et al. Molecular determinants of enterotoxigenic Escherichia coli heat-stable toxin secretion and delivery. Infect. Immun. https://doi.org/10.1128/IAI.00526-18 (2018).
Sack, D. A., Huda, S., Neogi, P. K., Daniel, R. R. & Spira, W. M. Microtiter ganglioside enzyme-linked immunosorbent assay for vibrio and Escherichia coli heat-labile enterotoxins and antitoxin. J. Clin. Microbiol. 11, 35–40 (1980).
Tsang, V. C., Wilson, B. C. & Maddison, S. E. Kinetic studies of a quantitative single-tube enzyme-linked immunosorbent assay. Clin. Chem. 26, 1255–1260 (1980).
Roy, K., Hamilton, D. J., Munson, G. P. & Fleckenstein, J. M. Outer membrane vesicles induce immune responses to virulence proteins and protect against colonization by enterotoxigenic Escherichia coli. Clin. Vaccin. Immunol. 18, 1803–1808 (2011).
Sixma, T. K. et al. Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J. Mol. Biol. 230, 890–918 (1993).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
J.M.F. was supported by funding from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) R01 AI126887, R01 AI089894, U01 AI095473; and the Department of Veterans Affairs (5I01BX001469-05). Research conducted by AS was also supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number T32AI007172. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. H.M.S. was supported by funding from the Department of Veterans Affairs (I01BX001142) and the National Institutes of Health (DK5606 and AA018071). HMS is the recipient of a Senior Research Career Scientist award # IK6BX006189.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, or the Department of Veterans Affairs.
Author information
Authors and Affiliations
Contributions
A.S., B.T., T.J.V., S.S., R.D.S., and W.B., performed experiments. J.C.M., B.A.R., M.M., C.S. E.T., and R.D.H., analyzed data. H.M.S., M.M., and J.M.F. planned experiments and wrote the manuscript
Corresponding author
Ethics declarations
Competing interests
R.D.H. and C.S. may receive royalty income based on the CompBio technology developed by R.D.H. and licensed by Washington University to PercayAI. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Declan McCole, V Viswanathan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sheikh, A., Tumala, B., Vickers, T.J. et al. Enterotoxigenic Escherichia coli heat-labile toxin drives enteropathic changes in small intestinal epithelia. Nat Commun 13, 6886 (2022). https://doi.org/10.1038/s41467-022-34687-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-34687-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
