
Abstract
Ca2+ influx through high-voltage-activated calcium channels (HVACCs) controls diverse cellular functions. A critical feature enabling a singular signal, Ca2+ influx, to mediate disparate functions is diversity of HVACC pore-forming α1 and auxiliary CaVβ1–CaVβ4 subunits. Selective CaVα1 blockers have enabled deciphering their unique physiological roles. By contrast, the capacity to post-translationally inhibit HVACCs based on CaVβ isoform is non-existent. Conventional gene knockout/shRNA approaches do not adequately address this deficit owing to subunit reshuffling and partially overlapping functions of CaVβ isoforms. Here, we identify a nanobody (nb.E8) that selectively binds CaVβ1 SH3 domain and inhibits CaVβ1-associated HVACCs by reducing channel surface density, decreasing open probability, and speeding inactivation. Functionalizing nb.E8 with Nedd4L HECT domain yielded Chisel-1 which eliminated current through CaVβ1-reconstituted CaV1/CaV2 and native CaV1.1 channels in skeletal muscle, strongly suppressed depolarization-evoked Ca2+ influx and excitation-transcription coupling in hippocampal neurons, but was inert against CaVβ2-associated CaV1.2 in cardiomyocytes. The results introduce an original method for probing distinctive functions of ion channel auxiliary subunit isoforms, reveal additional dimensions of CaVβ1 signaling in neurons, and describe a genetically-encoded HVACC inhibitor with unique properties.
Similar content being viewed by others


Introduction
Ion channels are integral membrane protein complexes that control flux of ions into and out of cells and organelles to regulate essential physiological processes including neuronal firing and synaptic transmission, muscle contraction, hormonal secretion, fluid homeostasis, and gene expression1. Surface membrane ion channels are often multi-subunit assemblies composed of pore-forming polypeptides assembled with auxiliary subunits. Within an ion channel class, functional diversification is frequently achieved by molecular diversity of both pore-forming α1 and auxiliary subunits in the form of either subtypes encoded by distinct genes or splice variants. Selective inhibition of distinct ion channels is a vital capability that not only enables delineation of their specific physiological roles but also has provided therapeutics for many diseases. Posttranslational inhibition of ion channels is typically accomplished pharmacologically using small molecules or peptides that target pore-forming α1 subunits. By contrast, posttranslational inhibition of ion channels based on the identity of their auxiliary subunits is rare, particularly when these accessory proteins are cytosolic. This deficiency is a critical blind spot that hampers in-depth understanding of the functional significance of ion channel molecular diversity and limits opportunities for developing novel potential therapeutics.
High-voltage activated calcium channels (HVACCs) convert information encoded in electrical signals to Ca2+ influx into cells to drive important biological responses including synaptic transmission in neurons and muscle contraction in the heart2. There are seven distinct HVACCs (CaV1.1-CaV1.4 CaV2.1- CaV2.3), classified based on the identity of the pore-forming α1 subunit (α1A- α1F; α1s), which contains the voltage sensor, selectivity filter, and channel pore. Mature HVACCs are multi-subunit complexes comprised of pore-forming α1 polypeptides assembled with auxiliary β, α2δ, and γ subunits. There are four CaVβ subunits isoforms (CaVβ1 - CaVβ4) encoded by distinct genes3. In most cell types, CaVβs are required for α1-subunit targeting to the plasma membrane4,5,6,7,8,9,10. Beyond surface density, CaVβs also regulate different aspects of channel gating including open probability (Po), voltage-dependence of activation and inactivation, and inactivation kinetics3,11,12,13,14,15,16.
HVACCs are important therapeutic targets for serious cardiovascular and neurological diseases2,17. There are selective small molecule and peptide toxin blockers that target HVACC pore-forming α1 subunits which have been critical in deciphering the distinctive physiological roles of α1 isoforms2,18,19,20. By contrast, there is no method currently available to inhibit HVACCs posttranslationally based on the identity of the CaVβ isoform associated with them. This limits the ability to discern the functional logic for CaVβ molecular diversity in tissues where multiple α1 and β-subunit isoforms are co-expressed. Here, we describe a nanobody, nb.E8, that exclusively binds auxiliary CaVβ1 subunits, and significantly inhibits whole-cell current through CaVβ1-associated HVACCs by decreasing channel surface density and Po. Fusing Nedd4L HECT domain to nb.E8 generated a tool we have named Chisel-1 (calcium channel inhibitor via selective targeting of CaVβ1) that selectively abolishes CaVβ1-bound HVACC currents in heterologous and native cells. Deploying Chisel-1 in hippocampal neurons revealed a dominant contribution of CaVβ1-associated HVACCs to excitation-transcription coupling in neurons. Overall, this work demonstrates a method for developing auxiliary-subunit-isoform-selective posttranslational inhibitors of HVACCs that can be broadly applied to other ion channels and multi-subunit membrane protein complexes.
Results
Rationale for developing post-translational CaVβ-isoform-selective HVACC inhibitors
The physiological roles of CaVβ isoforms have traditionally been probed using gene knockout or knockdown methods21,22,23,24,25. While such approaches have been invaluable in increasing understanding of the functional role of CaVβ molecular diversity in cells, they often lead to ambiguous results that can confound interpretation. Figure 1 shows two examples of how gene knockout/knockdown of CaVβ isoforms may yield results that are ambiguous to interpret. The first example stems from the observation that inducible knockout of CaVβ2 in adult mouse heart results in only a moderate impact on basal CaV1.2 current (ICa,L) amplitude26 (Fig. 1a). This result indicated that in contrast to the dogma established by heterologous expression experiments, plasma membrane targeting of CaV1.2 in adult heart cells was not absolutely dependent on association with a CaVβ subunit, leading to a quandary in interpretation. Is there a prominent subset of CaV1.2 channels in adult cardiomyocytes that are not complexed to CaVβ? The import of this question was heightened by the finding that association with a CaVβ is necessary for β-adrenergic upregulation of CaV1.2 which is necessary for the physiologically crucial fight-or-flight response27. We developed a nanobody-based molecule that posttranslationally inhibits voltage-gated Ca2+ channels by targeting auxiliary CaVβ subunits28. The molecule, termed CaV-aβlator, consists of a nanobody (nb.F3) that indiscriminately binds all four CaVβ isoforms, and is fused to the catalytic HECT domain of the E3 ubiquitin ligase, Nedd4L. CaV-aβlator potently inhibits CaV1/CaV2 channels by binding associated CaVβs and increasing ubiquitination of both α1 and CaVβ subunits thereby removing the channel complex from the cell surface. Expression of CaV-aβlator in cardiac myocytes eliminated ICa,L by retaining CaV1.2 in intracellular organelles, primarily late endosomes28, definitively revealing that essentially all CaV1.2 α1C subunits are bound to CaVβ in adult ventricular cardiomyocytes (Fig. 1a).

a Cartoon depicting hypothesized association of CaV1.2 with β2b subunits in adult ventricular myocytes (i) and the expected loss of β2b with gene knockout of this subunit (ii). Bottom, knock out of β2 in adult heart has only minimal impact on whole-cell Ca2+ current making it ambiguous whether most CaV1.2 are associated with β2b in adult ventricular cardiomyocytes. Posttranslational inhibition of cardiac CaV1.2 using a CaVβ-targeted nanobody fused to Nedd4L HECT (CaV-aβlator) domain eliminates CaV1.2 complexes from the membrane and abolishes current (iii), proving that CaV1.2 is stably associated with β2 in adult cardiomyocytes. b Schematic of a scenario where distinct CaVβ isoforms preferentially associate with particular α1-subunit types to mediate different functions (i). Knockdown of one β-isoform may lead to β reshuffling that lessens the functional impact of elimination of the particular CaVβ subunit (ii). By contrast, targeted posttranslational inhibition of the channel complex based on the β isoform would yield a qualitatively different result that more accurately reflects the functional logic of CaVβ molecular diversity in the cell (iii).
A second scenario visualizes a situation in which two CaVβ isoforms (e.g. CaVβ1 and CaVβ2) preferentially associate with distinct α1 subunits to mediate different downstream functional effects (outputs 1 and 2, Fig. 1b) in a cell. Gene knockout or knockdown of CaVβ1 isoform in this scenario may not result in an elimination of functional output 1 if CaVβ2 can replace it and has an overlapping function. This circumstance would result in an erroneous conclusion that CaVβ1 is not involved in output 1 (Fig. 1b). By contrast, if it were possible to selectively inhibit CaVβ1-associated channels post-translationally this would reveal a more accurate picture of the functional organization of CaV1/CaV2 channels and the divergent functions enabled by CaVβ molecular diversity.
Given the success of CaV-aβlator as a CaVβ-targeted potent posttranslational inhibitor of CaV1/CaV2 channels, we wondered whether it would be possible to develop CaVβ-isoform-selective blockers using a similar targeted ubiquitination design principle. Accordingly, we aimed to identify a nanobody that by contrast to nb.F3 was selective for a particular CaVβ isoform.
Isolation and characterization of a CaVβ1-selective nanobody
We previously immunized a llama with purified CaVβ1 and CaVβ3 subunits and generated a phagemid library of nanobodies (>1 × 107 library size) from peripheral blood mononuclear cells. Subsequent phage display using CaVβ1 as bait yielded a number of positive nanobody binders as determined by enzyme linked immunosorbent assay (ELISA). Several clones that showed strong binding by ELISA were sequenced and cloned into mammalian expression vectors28. To develop a robust and relatively high throughput approach to evaluate the binding and selectivity of the isolated nanobodies for CaVβ isoforms we adapted a flow cytometry Förster resonance energy transfer (flow-FRET) method29,30. As a test of the approach, we first evaluated the binding of nb.F3, which we previously established binds all four CaVβ isoforms using a low throughput membrane co-translocation assay28. We tagged nb.F3 with Venus and CaVβ isoforms (CaVβ1 - CaVβ4) with Cerulean fluorescence proteins, respectively. HEK293 cells were transiently co-transfected with Venus-nb.F3 and Cerulean-β or Cerulean alone as a negative control (Fig. 2a). In accord with our previous finding, Venus-nb.F3 showed strong FRET with all four Cerulean-tagged CaVβ isoforms (Fig. 2b). Scatter plots of FRET signal as a function of free Venus-nb.F3 (acceptor) concentration were fit with a 1:1 binding model yielding estimates of the relative binding affinity for all four CaVβs (Fig. 2b).

a Schematic of fluorescence resonance energy transfer (FRET) pair, Venus-nb.F3 + Cerulean-CaVβ. b Flow cytometry FRET (flow-FRET) scatter plots with FRET efficiency (ED) plotted vs free nb.F3 concentration. The blue lines fit a 1:1 binding model to the data. Data show nb.F3 binds all four CaVβ subunit isoforms. Kd,EFF, effective dissociation constant. c Schematic of Venus-nb.E8 + Cerulean-CaVβ. d Flow-FRET scatter plots and binding curves show nb.E8 exclusively binds CaVβ1. e Top, schematic of CaVβ structural organization. Conserved src homology 3 (SH3, orange) and nucleotide kinase (NK, green) domains are bounded by variable N- and C-termini, and separated by a HOOK domain. Bottom, cryoEM structure of CaVβ1a (PDB: 5GJV). f Flow-FRET scatter plots and binding curves for Cerulean-nb.E8 co-expressed with Venus-tagged β1b[NT-SH3-HOOK] (left), β1b[NK-CT] (middle), and β1b[SH3-HOOK] (right). g Alignment of region within SH3 domains of human CaVβ subunits. Highlighted region is a 21-residue sequence (η1) that shows some variability amongst CaVβ isoforms. h Flow-FRET scatter plots and binding curves for Cerulean-nb.E8 co-expressed with Venus-tagged chimeras β3[ηβ1b] (left) and β1b[ηβ3] (right). i Kd,EFF values. Data are means ± SEM. n = 17,409 cells over 7 independent experiments (grey bar), n = 8881 cells over 4 independent experiments (blue bar), n = 15,099 cells over 2 independent experiments (pink bar), and n = 6487 cells over 3 independent experiments (yellow bar). Source data are provided as a Source Data file.
We applied the flow-FRET approach to screen for binding and potential selectivity of different nanobody clones for distinct CaVβ isoforms. One of these, nb.E8, displayed strong binding to CaVβ1 with an affinity similar to that of nb.F3, but essentially no association with CaVβ2, CaVβ3, or CaVβ4 (Fig. 2c, d). To gain insights into the basis for the apparent selectivity of nb.E8 for β1, we probed for the structural determinants on CaVβ1 necessary for nb.E8 binding. CaVβs share a common architecture in which conserved SH3 and NK domains are separated by a variable HOOK region, and flanked by variable N and C-termini (V1 and V2, respectively)31,32,33 (Fig. 2e). We first divided CaVβ1 into two modules− NT-SH3-HOOK-SH3[β5] (with the essential SH3 β5 strand following the HOOK domain) and NK-CT− and tagged both moieties with Cerulean. Flow-FRET revealed that nb.E8 bound NT-SH3-HOOK-SH3[β5] but not NK-CT (Fig. 2f). CaVβs differ in their N-termini, with CaVβ1 having an extended N-terminus, raising expectations that this region might hold the essential determinants for selective nb.E8 binding. However, a construct with the CaVβ1 N-terminus deleted, SH3-HOOK-SH3[β5] retained robust binding to nb.E8, ruling out an important role of this region for the association (Fig. 1f). We reasoned that the SH3 domain was more likely to bind nb.E8 than the disordered HOOK region and thus searched for sequence variations among CaVβ subunits in the conserved SH3 domain. We identified a region on β1b SH3 domain encompassing a 310 helix (ƞ1) that contains several unique residues compared to other β isoforms (Fig. 2g). A chimeric construct, replacing the 310 helix of CaVβ3 with the equivalent regions from β1 (i.e., CaVβ3[ƞβ1]) was sufficient to reconstitute strong binding to nb.E8 (Fig. 2h, i). Conversely, the reverse chimera, CaVβ1b[ƞβ3], in which the ƞ1 region of β1b was replaced with that of β3 displayed a strongly reduced binding affinity to nb.E8 (Fig. 2h). The residual binding of CaVβ1b[ƞβ3] to nb.E8 suggested other determinants contribute to the high binding affinity of the CaVβ1/nb.E8 interaction. We turned to X-ray crystallography to gain more in-depth structural insights into how nb.E8 selectively binds CaVβ1, and how this contrasts with nb.F3 which non-selectively binds all four CaVβ subunits.
Structural basis for nb.F3 and nb.E8 association with CaVβ subunits
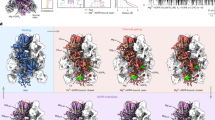
To understand the differential nanobody recognition modes, we crystalized CaVβ complexes containing the non-selective nanobody nb.F3 and the CaVβ1b-selective nanobody nb.E8 with two target CaVβs, CaVβ2a34 and CaVβ1b, and determined their structures using X-ray crystallography. CaVβ2a was crystallized as a 1:1 complex with nb.F3 (Supplementary Fig. 1a), whereas CaVβ1b was crystallized as a 1:1:1 complex with nb.F3 and nb.E8 (Supplementary Fig. 1b). Both complexes diffracted X-rays to 2.0 Å resolution and were solved by molecular replacement (Supplementary Fig. 1c, d; Supplementary Table 1). The structures show that both nanobodies primarily target the CaVβ SH3 domain but do so by binding to opposite sides of the structure (Fig. 3a, b).

a Cartoon diagram of the nb.F3:CaVβ2a complex. CaVβ2a SH3 (helices (dark green) and β-strands (light green)) and NK (helices (dark blue) and β-strands (cyan)) domains are indicated. Nb.F3 (yellow) is shown with a semi-transparent surface. Nb.F3 Site 1 and Site 2 binding regions are indicated by the red and orange ovals, respectively. b Cartoon diagram of nb.F3:nb.E8:CaVβ1b complex. SH3 (helices (green) and β-strands (lime)) and NK (helices (purple) and β-strands (violet)) domains are indicated. Nb.F3 (yellow) and nb.E8 (yellow orange) are shown with a semi-transparent surface. Site 1 and Site 2 binding regions are indicated by the red and orange ovals, respectively. Light green oval denotes nb.E8 binding site. Lower panel shows view from the SH3 domain N-terminus as indicated by the arrows. ABP indicates AID binding pocket location in (a) and (b), (c) View of nb.F3:CaVβ2a Site 1 interactions. d View of nb.F3:CaVβ2a Site 2 interactions. e View of nb.E8:CaVβ1b interactions. Red ovals indicate CaVβ sites that differ among isoforms. Complementarity determining regions (CDRs) are colored similarly in all panels: CDR1 (purple), CDR2 (red), CDR3 (magenta). In (c–e) nanobody residues are in black, CaVβ residues are labeled in grey italics.
In the nb.F3:CaVβ2a complex, nb.F3 displays the canonical nanobody architecture (Supplementary Fig. 1e, f) and CaVβ2a is largely unchanged from its AID-bound form35 with the exception of a displacement of the SH3 α2 helix and the resolution of the 263–283 loop spanning the η2 and α4 NK domain helices and ten residues of the C-terminal V3 domain (Root mean squared deviation Cα, RMSDCα 0.568 Å) (Supplementary Fig. 1g). The nb.F3:CaVβ2a complex showed that nb.F3 engages CaVβ2a using two interaction sites encompassing ~forty nb.F3 residues (Fig. 3a; Supplementary Fig. 2a, b). Site 1 comprises interactions with the three complementarity determining regions (CDRs) CDR1, CDR2 and CDR3, the majority of which are made between nb.F3 CDR3 with Phe62, Lys98, Asn 101, and Trp104 from β1, β2, and β3 of the SH3 core and CDR2 with Tyr200, Asp221, and Val222 from the SH3 β5 strand contributed from the NK domain (Fig. 3c; Supplementary Fig. 2a). Site 1 buries 866 Å2 and contains a mixture of hydrogen bonding and van der Waals interactions (Supplementary Fig. 2a). Remarkably, the CDR3 Tyr102-Trp103 segment binds a small SH3 domain surface pocket made by Phe62, Trp104, and Val222 previously identified as a potential protein–protein interaction site by virtue of crystallographically observed binding of the short CaVβ2a V2/HOOK domain sequence, Arg207-Phe210 (RMPF) to this site31,36. Structural comparison shows that CDR3 Tyr102-Trp103 and the RMPF peptide make remarkably similar interactions even though their peptide chains run in opposite directions (Supplementary Fig. 3a). This observation highlights the potential of this site as a protein-protein interaction surface. In contrast to Site1, Site 2 is smaller (513.1Å2) and rather than CDR interactions, comprises interactions of nb.F3 framework strands C”, D, and E residues with the 275-295 loop located between the η2 and α4 NK domain elements. Notably, this loop is disordered in other CaVβ2a structures31,35 (Supplementary Fig. 1f).
Nb.F3 also uses Sites 1 and 2 to bind CaVβ1b, and the way nb.F3 binds to CaVβ1b in the nb.F3:nb.E8:CaVβ1b complex is very similar to nb.F3:CaVβ2a components (RMSDCα = 0.731 Å) (Supplementary Fig. 3b). Overall, the conformations and interactions of Site 1 are conserved and bury a similar amount of surface area (869 Å2). CDR3 makes the same interactions as in the nb.F3:CaVβ2a complex (Supplementary Fig. 3c), while there are small differences in the interactions with CDR2 (Supplementary Fig. 2b). Although Site 2 in the CaVβ1b complex remains primarily a site involving framework residues, there are differences from nb.F3:CaVβ2a. Most notably, there is an absence of interactions with the equivalent of the CaVβ2a 275–295 loop; as a consequence, Site 2 buries much less surface area (146 Å2). Most of residues in Site 1 and Site 2 are highly conserved amongst different CaVβs, explaining the relative lack of specificity for nb.F3.
The nb.F3:nb.E8:CaVβ1b complex shows that the β1 selective nanobody nb.E8, which has a longer CDR3 than nb.F3, also has the canonical nanobody structure (Supplementary Fig. 3e) and uses its three CDRs to bind to the CaVβ1b SH3 domain face formed by the long loop between SH3 domain strands β1 and β2 opposite to the nb.F3 epitope (Fig. 3b). This interaction buries 853Å2 and involves a mixture of hydrogen bonds, salt bridges, and van der Waals interactions (Supplementary Fig. 2c). One notable interaction is made by the Val103-Arg104 portion of CDR3. This loop wedges between the SH3 domain α2 helix and η1 loop allowing Arg104 to make a salt bridge with Asp117 and a series of backbone mediated hydrogen bonds to Asn112 and Gln172 of CaVβ1b, matching the region identified by our chimera studies (Fig. 2g, h), and thereby defining the positions that are important for the selectivity of nbE8 for CaVβ1 over other isoforms. These include residues in the center of the binding interface, Gly110, and Asn112 (Fig. 3e), and variations at Gly116 where other CaVβ isoforms have larger residues that would likely interfere with the nb.E8 Arg104 interactions (Fig. 2g; Supplementary Fig. 4). Hence, together the structural data define modes for both non-selective (nb.F3) and selective (nb.E8) recognition of CaVβ that exploit different aspects of the SH3 domain structure.
Nb.E8 selectively inhibits recombinant CaV1/CaV2 channels reconstituted with CaVβ1
We evaluated whether nb.E8 (expressed in a P2A-CFP plasmid vector) affected HVACC functional expression by transient co-expression with recombinant human CaV2.2 (α1B + β + α2δ−1) channels reconstituted in HEK293 cells (Fig. 4). Contemporaneous control experiments used HEK293 cells co-expressing CFP and recombinant CaV2.2 channels. We used path-clamp electrophysiology to record whole-cell currents through reconstituted CaV2.2 channels using 5 mM Ba2+ as charge carrier. Control CaV2.2 channels reconstituted with any of the four CaVβ isoforms yielded robust whole-cell currents that activated at a threshold between −15 and −10 mV, and peaked at +10 mV (Fig. 4). CaV2.2 channels reconstituted with β1 and nb.E8 yielded several differences in channel properties compared to control. First, the whole-cell current amplitude was strongly decreased (Ipeak = −102.8 ± 15.78 pA/pF, n = 20 for CFP; and Ipeak = −22.42 ± 4.321 pA/pF, n = 16 for nb.E8, P = 6.5 × 10−5, unpaired two-tailed Student’s t test) (Fig. 4a, b). Second, the voltage-dependence of channel activation was right-shifted as indicated by a + 10-mV shift in the V0.5 of the tail activation curve (Supplementary Fig. 5). Third, channels expressed with nb.E8 displayed a faster rate of voltage-dependent inactivation (VDI) that was evident in exemplar traces (Fig. 4a), and quantified in population data as a decrease in the fractional current remaining after 20 ms (r20 = I20/Ipeak) across a range of test pulse voltages (r20 at +10 mV = 0.96 ± 0.08, n = 20 for CFP; and r20 at +10 mV = 0.62 ± 0.05, n = 13 for nb.E8, P = 7.4 × 10−5, unpaired two-tailed Student’s t test) (Fig. 4c). In sharp contrast, in CaV2.2 channels reconstituted with β2, β3, or β4, co-expressing nb.E8-P2A-CFP produced no change in current amplitude or gating behavior compared to control (Fig. 4d–l).

a Exemplar family of whole cell currents from HEK293 cells expressing reconstituted CaV2.2 channels (α1B+β1b+α2δ-1) in the absence (left) or presence (right) of nb.E8. b Population current density vs voltage (J-V) relationship for cells expressing α1B+β1b+α2δ-1 in the absence (black circles; n = 20 cells examined over 4 independent experiments) or presence (red circles; n = 16 cells examined over 4 independent experiments) of nb.E8. *P = 6.5 × 10−5, two-tailed unpaired t-test. c Fractional current remaining after 20 ms (r20) at various test pulse voltages for cells expressing α1B+β1b+α2δ−1 in the absence (black circles; n = 20 cells examined over 4 independent experiments) or presence (red circles; n = 13 cells examined over 4 independent experiments) of nb.E8. *P = 7.4 × 10−5, two-tailed unpaired t test. d–f Data for α1B+β2b+α2δ−1 ± nb.E8, same format as (a–c). e n = 8 cells examined over 3 independent experiments (black symbols) and n = 6 cells examined over 3 independent experiments (red symbols). f n = 7 cells examined over 3 independent experiments (black symbols) and n = 5 cells examined over 3 independent experiments (red symbols). g–i Data for α1B+β3+α2δ−1 ± nb.E8, same format as a–c. h n = 17 cells examined over 4 independent experiments (black symbols) and n = 7 cells examined over 3 independent experiments (red symbols). (i) n = 12 cells examined over 4 independent experiments (black symbols) and n = 6 cells examined over 3 independent experiments (red symbols). j–l Data for α1B+β4+α2δ−1 ± nb.E8, same format as a–c. k n = 7 cells examined over 3 independent experiments (black symbols) and n = 8 cells examined over 3 independent experiments (red symbols). l n = 12 cells examined over 3 independent experiments (black symbols) and n = 10 cells examined over 3 independent experiments (red symbols). Data are means ± SEM. Source data are provided as a Source Data file.
We investigated whether nb.E8 could selectively inhibit other CaV1/CaV2 channel isoforms as long as they were reconstituted with β1 subunit. Indeed, we found that nb.E8 significantly inhibited CaV2.1, CaV2.3, and CaV1.3 channels that were reconstituted with β1b but not β2b, β3, or β4 subunits. Thus, the ability of nb.E8 to selectively inhibit β1-bound channels is a general feature for CaV1/CaV2 channels.
Nb.E8 inhibits surface density and P o of CaV2.2 channels reconstituted with CaVβ1
The whole-cell current (I) is related to microscopic channel properties by the relation I = N × i × Po, where N is the total number of channels at the cell surface, i is the unitary current amplitude, and Po is the single-channel open probability. We first determined whether a decrease in N contributes to the impact of nb.E8 on I. To address this, we applied a flow cytometry-based assay to measure surface density of CaV2.2 channels; the method utilizes an engineered α1B that harbors a tandem repeat of the high-affinity α-bungarotoxin-binding site (BBS) (derived from the nicotinic acetylcholine receptor) in the extracellular domain IV S5-S6 loop6,7. We co-expressed BBS-α1B with β1b-YFP and α2δ−1 to simultaneously measure surface (Alexa-647 conjugated α-bungarotoxin) and total β1b (YFP fluorescence) levels in non-permeabilized HEK293 cells using flow cytometry (Fig. 5a, b; Supplementary Fig. 6). Compared to control cells in which CFP was co-expressed with BBS-α1B + β1b-YFP + α2δ−1, expression of nb.E8 significantly decreased CaV2.2 surface density (50% reduction) while only moderately affecting β1b expression (~20% reduction in YFP fluorescence) (Fig. 5b, c).

a Schematic of BBS-α1B + β1b-YFP ± nb.E8. b Flow cytometry contour plots showing CaV2.2 surface density (Alexa647) vs β1b expression (YFP) in HEK293 cells expressing α1B+β1b+α2δ−1 with either CFP (left) or nb.E8 (right). c Bar charts showing impact of nb.E8 on CaV2.2 surface density (n = 6 over 3 independent experiments for both CFP and nb.E8 groups) and β1b-YFP expression (n = 6 over 3 independent experiments for both CFP and nb.E8 groups). *P = 0.0052 compared to CFP control, two-tailed unpaired t test. d Schematic of cell-attached single channel recording. e Exemplar single channel recordings evoked by slow ramp protocols in HEK293 cells expressing α1B+β1b+α2δ−1 with either CFP (left) or nb.E8 (right). f Ensemble average open probability vs voltage (Po-V) relationships in cells expressing α1B+β1b+α2δ−1 with either CFP (gray) or nb.E8 (red). g Bar charts showing impact of nb.E8 on maximal single-channel open probability, Po,max (n = 5 over 3 independent experiments for CFP, and n = 8 over 3 independent experiments for nb.E8). *P = 0.0017 compared to CFP control, two-tailed unpaired t test. Data are means ± SEM. Source data are provided as a Source Data file.
To determine whether changes in i and/or Po contribute to nb.E8-mediated decrease in I we turned to single-channel recordings which enable direct measurement of these parameters (Fig. 5d). We used 40 mM Ba2+ as charge carrier in the cell-attached patch clamp mode and a slow-voltage ramp protocol to obtain stochastic channel openings, which reflect near-steady state Po at each voltage12,37. Control CaV2.2 channels (α1B + β1b + α2δ−1) that were expressed with CFP had robust openings with a maximal Po (Po,max) of 0.35, while channels co-expressed with nb.E8 exhibited a Po,max that was approximately half of the control (Po,max = 0.35 ± 0.032, n = 5 for CFP; Po,max = 0.16 ± 0.029, n = 8 for nb.E8, P = 0.0017 unpaired two-tailed Student’s t test) (Fig. 5e–g). The unitary current amplitude was unchanged. Overall, these data indicate that nb.E8 selectively inhibits β1b-bound CaV2.2 channels by decreasing both N and Po.
Nb.E8-HECTNedd4L (Chisel-1) selectively ablates β1-associated HVACC currents
Though nb.E8 intrinsically acts as a potent inhibitor of β1-associated HVACCs the elimination of current is incomplete (Fig. 4a, b). We previously showed that fusing the HECT domain of Nedd4L to nb.F3 created a construct, CaV-aβlator, that completely eliminated HVACCs irrespective of the β-subunit isoform with which they were reconstituted28. The mechanism involved CaV-aβlator targeting to auxiliary β subunits in channel complexes and producing a ubiquitination dependent intracellular relocation of the channel complex. We wondered whether similar engineering of nb.E8 would generate an equally potent, but β1-selective, HVACC inhibitor. Accordingly, we created a chimeric nb.E8-HECTNedd4L (Chisel-1) construct downstream of CFP-P2A to permit separate expression of reporter fluorescence and engineered nb.E8 proteins, respectively (Fig. 6a). When co-expressed with α1C + β1b, Chisel-1 decreased expression of both β1b and α1C, most likely due to ubiquitin-mediated degradation, as supported by the increased ubiquitination of both subunits (Fig. 6c–f). Whole-cell patch clamp recordings revealed that Chisel-1 essentially eliminated all current through recombinant CaV2.2 channels reconstituted with β1b (Jpeak = −102.8 ± 15.78 pA/pF, n = 20 for CFP; and Jpeak = −1.65 ± 0.34 pA/pF, n = 10 for Chisel-1, P = 3.82 × 10−6, unpaired two-tailed Student’s t test) (Fig. 6g, h). In sharp contrast, Chisel-1 had no effect on CaV2.2 channels reconstituted with β2, β3, or β4 subunits (Fig. 6i–k). We obtained similar results for recombinant CaV2.1, CaV2.3, and CaV1.3 channels— Chisel-1 abolished current through these channels when they were reconstituted with β1b, but was completely ineffective when the channels were formed with β2, β3, or β4 (Fig. 7).

a Schematic showing modular structures of Nedd4L and Chisel-1. b Schematic of α1C + β1b ± Chisel-1. c Left, β1b immunoblot in untransfected cells (UT), or cells transiently transfected with α1C + β1b and CFP, nb.E8 or Chisel-1. Right, Bar charts showing impact of Chisel-1 on normalized β1b expression (n = 5 independent experiments each for CFP, nb.E8, and Chisel-1 groups). d Left, β1b pulldown followed by β1b immunoblot. Middle, β1b pulldown followed by ubiquitin immunoblot. Right, Bar chart showing Chisel-1 enhances ubiquitination of β1b (n = 5 independent experiments each for CFP, nb.E8, and Chisel-1 groups). e Left, α1C immunoblot. Right, impact of Chisel-1 on α1C normalized expression (n = 6 independent experiments each for CFP, nb.E8, and Chisel-1 groups). f Left, α1C pulldown followed by α1C immunoblot. Middle, α1C pulldown followed by ubiquitin immunoblot. Right, Bar chart showing Chisel-1 enhances ubiquitination of α1C (n = 6 independent experiments each for CFP, nb.E8, and Chisel-1 groups). g Exemplar family of whole-cell currents in HEK293 cells expressing α1B+β1b+α2δ−1 in the absence (left) or presence (right) of Chisel-1. h Population current density vs voltage (J-V) relationship for cells expressing α1B+β1b+α2δ−1 in the absence (black circles; n = 20 over 5 independent transfections) or presence (red circles; n = 10 over three independent transfections) of Chisel-1. i J-V relationship for α1B+β2b+α2δ−1 ± Chisel-1, same format as (h). (For CFP, n = 8 over 3 independent transfections, black symbols; for Chisel-1, n = 6 over 3 independent transfections, red symbols). j J-V relationship for α1B+β3+α2δ−1 ± Chisel-1, same format as (h) (For CFP, n = 17 over 3 independent transfections, black symbols; for Chisel-1, n = 8 over 3 independent transfections, red symbols). k J-V relationship for α1B+β4+α2δ−1 ± Chisel-1, same format as (h) (for CFP, n = 7 over 3 independent transfections, black symbols; for Chisel-1, n = 10 over 3 independent transfections, red symbols). Data are means ± SEM. Source data are provided as a Source Data file.

a Population current density vs voltage (J-V) relationship for cells expressing α1A+β+α2δ−1 and co-expressed with CFP (black symbols), nb.E8 (cyan symbols) or Chisel-1 (red symbols): α1A+β1b+α2δ−1 (CFP, n = 6; nb.E8, n = 6, Chisel-1, n = 3); α1A+β2b+α2δ−1 (CFP, n = 7; nb.E8, n = 6, Chisel-1, n = 5); α1A+β3+α2δ−1 (CFP, n = 11; nb.E8, n = 6, Chisel-1, n = 6); α1A+β4+α2δ−1 (CFP, n = 10; nb.E8, n = 5, Chisel-1, n = 8). b J-V relationship for cells expressing α1D+β+α2δ−1 with CFP (black symbols), nb.E8 (cyan symbols) or Chisel-1 (red symbols): α1D+β1b+α2δ−1 (CFP, n = 8; nb.E8, n = 9, Chisel-1, n = 5); α1D+β2b+α2δ−1 (CFP, n = 8; nb.E8, n = 8, Chisel-1, n = 7); α1D+β3+α2δ−1 (CFP, n = 3; nb.E8, n = 5, Chisel-1, n = 5); α1D+β4+α2δ−1 (CFP, n = 7; nb.E8, n = 10, Chisel-1, n = 4). c J-V relationship for cells expressing α1E+β+α2δ−1 with CFP (black symbols), nb.E8 (cyan symbols) or Chisel-1 (red symbols): α1E+β1b+α2δ−1 (CFP, n = 6; nb.E8, n = 8, Chisel-1, n = 6); α1E+β2b+α2δ−1 (CFP, n = 7; nb.E8, n = 5, Chisel-1, n = 5); α1E+β3+α2δ−1 (CFP, n = 8; nb.E8, n = 7, Chisel-1, n = 4); α1E+β4+α2δ−1 (CFP, n = 12; nb.E8, n = 5, Chisel-1, n = 6). Data are means ± SEM. Source data are provided as a Source Data file.
Flow cytometry surface staining assay showed that in cells co-expressing BBS-α1B + β1b-YFP Chisel-1 reduced the surface density of BBS-α1B beyond that achieved with nb.E8 (~60% decrease), while only moderately inhibiting β1b expression (Supplementary Fig. 7).
Probing Chisel-1 efficacy and selectivity in skeletal and cardiac muscle cells
We wondered whether Chisel-1 could potently and selectively inhibit endogenous β1-bound native HVACCs in primary cells. Skeletal and cardiac muscle cells provided ideal systems to address this question due to certain unique characteristics. First, they both have intricate cytoarchitectures with complicated intracellular environments specialized for their contractile function. This complex cytoplasmic milieu thus provides a good challenge for monitoring the efficacy of Chisel-1. Second, adult skeletal muscle fibers exclusively express CaV1.1 (α1S) in complex with β1a (Fig. 8a), whereas adult ventricular cardiomyocytes predominantly express CaV1.2 (α1C) in complex with β2 (Fig. 8e) with no contribution from β126,38. Thus, the efficacy of Chisel-1 can be determined by how effectively it inhibits CaV1.1 in skeletal muscle, while its selectivity can be deduced from how ineffective it is against CaV1.2 in ventricular cardiomyocytes.

a Schematic of skeletal muscle Cav1.1 complex. b Images of isolated flexor digitorum brevis (FDB) fibers either untransfected (top) or transfected with Chisel-1-P2A-CFP (bottom). c Top, exemplar whole-cell currents from isolated FDB fibers expressing CFP (left) or Chisel-1 (right). Bottom, Population J-V curves from isolated FDB fibers expressing CFP (black squares; n = 13 over 3 independent experiments) or Chisel-1 (red squares; n = 13 over 3 independent experiments). d Top, exemplar gating currents from isolated FDB fibers expressing CFP (left) or Chisel-1 (right). Bottom, Population Q-V curves from isolated FDB fibers expressing CFP (black circles; n = 7 over 2 independent experiments) or Chisel-1 (red circles; n = 8 over 2 independent experiments). *P = 9.32 × 10−5 compared to CFP control, two-tailed unpaired t test. e Schematic of ventricular cardiomyocyte CaV1.2 complex. f Confocal images of cardiomyocytes expressing mCherry (top) or Chisel-1-P2A-mCherry (bottom). g Population J-V curves from isolated ventricular myocytes expressing mCherry (black triangles; n = 13 over 3 independent experiments) or Chisel-1 (red triangles; n = 11 over 3 independent experiments). h Top, exemplar whole-cell currents from ventricular myocytes expressing mCherry (left) or Chisel-1 (right) before (black) and after (cyan) application of 1 μM forskolin. Bottom, lack of effect of Chisel-1 on forskolin induced increase in ICa,L in ventricular myocytes (mCherry, n = 14; Chisel-1, n = 5). Data are means ± SEM. Source data are provided as a Source Data file.
We performed in vivo electroporation of flexor digitorum brevis (FDB) fibers of adult mice with plasmids encoding either CFP (control) or Chisel-1 (expressed in a CFP-P2A cassette). Ten days after electroporation, muscle fibers were dissociated into individual cells many of which expressed CFP fluorescence, confirming transgene expression (Fig. 8b). Control skeletal muscle myocytes expressing CFP displayed large whole cell Ca2+ currents that were completely eliminated in cells expressing Chisel-1 (Ipeak = −12.97 ± 1.51 pA/pF, n = 13 for CFP; Ipeak = −0.02 ± 0.61 pA/pF, n = 13 for Chisel-1, P = 6.37 × 10−7) (Fig. 8c). Gating current measurements revealed Chisel-1 significantly decreased, but did not completely eliminate, intramembrane charge movement, consistent with a reduced CaV1.1 surface density compared to control cells expressing CFP (Qmax = 30.97 ± 1.75 pA/pF, n = 7 for CFP; Qmax = 15.47 ± 2.17 pA/pF, n = 8 for Chisel-1, P = 9.32 × 10−5) (Fig. 8d). Chisel-1 also reduced stimulus-evoked rhod2 Ca2+ transients by 50% compared to control skeletal muscle fibers expressing CFP (Supplementary Fig. 8), consistent with excitation-contraction coupling in skeletal muscle being mediated by voltage-induced Ca+2 release rather than the Ca2+-induced Ca2+ release found in heart cardiomyocytes39. These results demonstrate that Chisel-1 eliminates Ca2+ current through CaVβ1- associated CaV1.1 channels in skeletal muscle and this effect is partially mediated through a reduction in the channel surface density. The discrepancy between the near complete ablation of whole-cell current and the 50% reduction in CaV1.1 surface density suggests that Chisel-1 inhibits the Po of channels remaining at the surface even more strongly than we found for nb.E8. This could be due to the enhanced ubiquitination which has been shown to inhibit gating of some channels40.
In sharp contrast with the elimination of CaV1.1 current in skeletal muscle, adenovirus-mediated expression of Chisel-1 in adult guinea pig ventricular cardiomyocytes had no effect on whole-cell CaV1.2 functional expression (Ipeak = −7.08 ± 0.97 pA/pF, n = 15 for mCherry; Ipeak = −8.39 ± 1.33 pA/pF, n = 8 for Chisel-1) (Fig. 8f, g). Importantly, we previously showed that CaV-aβlator (comprised of nb.F3 fused to Nedd4L HECT domain) eliminates CaV1.2 current in adult guinea pig ventricular cardiomyocytes28. CaV1.2 in adult cardiac myocytes can target to the cell surface and support functional currents independent of CaVβ binding26. Thus, it was possible that Chisel-1 could interfere in some way with CaVβ functional interaction with CaV1.2 in cardiomyocytes that would, nevertheless, not register as a frank change in current amplitude. However, interaction of CaVβ with CaV1.2 is obligatory for up-regulation of cardiac ICa,L by activated PKA27, offering a path to test whether Chisel-1 disrupted α1C/β interaction in heart cells. We found no difference in the magnitude of forskolin-induced increase in ICa,L between control cardiomyocytes expressing mCherry (4.19 ± 0.32 fold) and cells expressing Chisel-1 (4.12 ± 0.66 fold) (Fig. 8h) indicating the α1C/β2b interaction remained intact. Overall, the sharp dichotomy of Chisel-1 effects on ICaL in skeletal versus cardiac myocytes demonstrates the efficacy of this unique tool in selectively eliminating current through CaVβ1-bound channels in native cells.
Chisel-1 inhibits excitation-transcription coupling in hippocampal neurons
Hippocampal neurons express multiple CaV channel α1 (including CaV1.2, CaV1.3, CaV2.1, CaV2.2, and CaV2.3) and CaVβ (CaVβ1-CaVβ4) subunits41,42,43,44,45, representing a context in which there is the bewildering possibility of at least 20 molecularly distinct CaV channel α1/β subunit combinations. Chisels that are able to selectively inhibit CaV channels based on the identity of their associated CaVβ isoform could be an exceptional tool to help discriminate the functional logic of CaVβ molecular diversity in neurons. Accordingly, we determined whether Chisel-1 would be effective in revealing physiological effects mediated by CaVβ1-bound CaV channels in cultured hippocampal neurons.
Lentiviral-mediated expression of mCherry-P2A-Chisel-1 resulted in a substantial reduction in CaVβ1 immunofluorescence as compared to control neurons expressing mCherry alone (Fig. 9a). Western blot confirmed the diminished expression of CaVβ1 in neurons expressing Chisel-1 (Fig. 9b). By contrast, Chisel-1 had no impact on expression levels of CaVβ2 or CaVβ3. However, neurons expressing Chisel-1 showed a higher expression of CaVβ4 compared to control (Fig. 9b). Interestingly, β4-null lethargic mice selectively displayed a compensatory increase in β1b expression46, suggesting a specific reciprocal regulation of these two auxiliary CaVβ isoforms.

a Representative hippocampal neuron cultures expressing mCherry (top row) or Chisel-1-P2A-mCherry (bottom row) with immunofluorescence detection of β1b (green) and MAP2 (purple). Images are representative of three independent experiments. b Representative CaVβ isoform immunoblots from hippocampal neurons expressing either mCherry or Chisel-1. Immunoblots are representative of three independent experiments. c Time course of mean 90 KCl-evoked Ca2+ transients in hippocampal neurons expressing either mCherry (black line) or Chisel-1 (red trace). d Bar charts showing impact of Chisel-1 on 90 KCl-evoked Ca2+ transients before and after the addition of CaV1/CaV2 channel blockers (mCherry, n = 20 over 3 independent experiments; Chisel-1, n = 30 over 3 independent experiments). e Top, schematic of experimental protocol for assessing excitation-transcription coupling in cultured hippocampal neurons. Bottom, exemplar images showing phosphoCREB (pCREB), microtubule associated protein 2 (MAP2), and DAPI staining of fixed mCherry-expressing hippocampal neurons exposed to either 5 or 40 mM KCl. NBQX, 2,3-dioxo-6-nitro-7-sulfamoyl-benzo[f]quinoxaline; TTX tetrodotoxin, AP5 2-Amino-5-phosphonopentanoic acid. f Bar chart showing 40 KCl-induced nuclear translocation of pCREB signal and its inhibition by a cocktail of CaV1/CaV2 channel inhibitors and Chisel-1: 5 K, n = 146; 40 K, n = 166; 40 K + nisoldipine + ω-conotoxin GVIA + ω-agatoxin, n = 139; Chisel-1, n = 189; CaV-aβlator, n = 48. * significantly different compared to 40 mM KCl condition by one-way ANOVA and Tukey’s multiple comparison test: 5 K, P = 0; 40 K + nisoldipine + ω-conotoxin GVIA + ω-agatoxin, P = 2.1 × 10−8; Chisel-1, P = 5.5 × 10−9; CaV-aβlator, P = 0. Data are means ± SEM. Source data are provided as a Source Data file.
Hippocampal neurons expressing Chisel-1 displayed a diminished KCl-evoked Ca2+ transient amplitude consistent with a decrease in whole-cell current (Fig. 9c, d). We examined whether CaVβ1-bound channels participate in excitation-transcription coupling in hippocampal neurons47. In neurons pre-treated with NBQX + AP5 + TTX, a 3-minute exposure to 40 mM KCl resulted in a 6-fold increase in phosphoCREB immunoreactivity in the nucleus, consistent with previous reports (Fig. 9e, f). The 40 KCl-induced increase in nuclear phosphoCREB was strongly reduced in the presence of a cocktail of CaV1/CaV2 channel blockers (nisoldipine + ω-conotoxin GVIA + ω-agatoxin IVA) fitting with the known role of VGCCs in this phenomenon (Fig. 9f). CaV-aβlator inhibited excitation-induced phosphoCREB signal in the nucleus of hippocampal neurons to a similar extent as the cocktail of CaV channel blockers (Fig. 9f). With these controls established, we discovered that hippocampal neurons expressing Chisel-1 also displayed a robust reduction in the 40 KCl-evoked increase in nuclear phosphoCREB staining, indicating that CaVβ1-bound channels play a prominent role in excitation-transcription coupling in hippocampal neurons (Fig. 9f).
Discussion
This work describes the development and application of Chisel-1, a unique tool that potently and selectively inhibits CaVβ1-bound HVACC in cells by promoting targeted ubiquitination of the channel complex, decreasing channel surface density (N), and reducing single-channel Po. The mechanism of action of Chisel-1 is qualitatively different from conventional gene knockout or shRNA knockdown approaches and yields complementary information to these established methods. More broadly, our success with Chisel-1 supplies a blueprint for developing Chisels that inhibit not only HVACCs based on the CaVβ isoform (β2 – β4) they are associated with, but also the myriad other multi-subunit ion channel complexes. Overall, this method significantly adds to the arsenal of tools available to probe physiological/pathophysiological roles of ion channel subunit molecular diversity, and to develop novel genetically-encoded ion channel inhibitors as research tools or potential therapeutics.
Ca2+ influx through HVACCs controls a rich variety of physiological responses in excitable cells. The diversified responses ensuing from a singular signaling event, Ca2+ influx, is mediated in part by molecular heterogeneity of HVACC pore-forming α1 and auxiliary CaVβ subunits. Selective small molecule or toxin blockers available for the different CaVα1 isoforms have been invaluable in delineating their distinctive physiological roles2. By contrast, there are no equivalent molecules or methods that can post-translationally eliminate HVACC function based on the identity of the associated CaVβ isoform. CaVβ1 - CaVβ4 subunits have some overlapping functions such as facilitating surface membrane targeting of co-expressed α1 subunits, shifting the voltage-dependence of channel activation in the hyperpolarizing direction, and increasing channel Po3. However, the individual β-subunit isoforms also confer distinctive rates of inactivation and steady-state inactivation properties to HVACCs that hint at unique functional roles in native cells3. Results from knockout mice and model organisms confirm the notion of unique physiological roles for different CaVβ isoforms. For example, CaVβ1 knockout mice die at birth due to asphyxia, a consequence of β1a being the sole CaVβ isoform expressed in skeletal muscle21. β1-null zebrafish (relaxed) are paralyzed owing to loss of skeletal muscle EC coupling48. Swapping in a different CaVβ isoform for CaVβ1a in either dysgenic mouse or β1-null zebrafish skeletal muscle restored whole cell CaV1.1 current but did not recover tetrad formation or EC coupling49, explicitly demonstrating the principle of both overlapping and unique functions of distinct CaVβ isoforms. Similarly, embryonic knockout of β2 is lethal due to impaired development of the heart and an absence of cardiac contractions23. Transgenic expression of β2 in the hearts of β2-null mice rescued viable animals which were, nevertheless, deaf24 and experienced visual impairments reminiscent of patients with congenital stationary night blindness22. The interpretation of functional effects of CaVβ knockouts in cell types such as neurons that express multiple CaVα1 and CaVβ isoforms can be more complicated. For example, CaVβ3 knockout mice displayed alterations in learning and memory tasks, yet showed no changes in whole-cell Ca2+ currents from hippocampal neurons50; and selectively exhibited a dampened response to pain initiated by chemical inflammation, but with no overt changes in HVA Ca2+ currents25. These conflicting results from CaVβ3 knockout studies likely reflect the phenomenon of CaVβ subunit reshuffling in which other CaVβ isoforms present in neurons bind to CaVα1 subunits and occupy slots that would otherwise have been engaged by CaVβ3. Post-translational inhibition of HVACCs based on their specific constituent CaVβ isoform as embodied in the Chisel concept would be expected to limit the confounding effects of CaVβ reshuffling and reveal the full scope of the functional logic of CaVβ molecular diversity. Consistent with this notion, Chisel-1 reduced the amplitude of high KCl-evoked Ca2+ transients and strongly inhibited excitation-transcription coupling in hippocampal neurons. These results provide an impetus to develop Chisels that are selective for CaVβ2, CaVβ3, and CaVβ4 isoforms as a focus for future studies. Beyond their utility as tools to decipher physiological roles of CaVβ isoforms, Chisels have potential therapeutic applications as genetically-encoded HVACC inhibitors with selectivity for distinct CaVβ isoforms. As an example of their potential advantages, we consider CaVβ3 knockout mice which display reduced anxiety but increased aggression51. There are distinctive neuronal circuits for the two types of behaviors suggesting the possibility it may be possible to reduce anxiety with localized expression of a Chisel-3 without inducing aggressive behaviors.
Our results are also noteworthy for providing the first proof of concept of using an unmodified CaVβ-targeted nanobody to inhibit current amplitude and modulate gating properties of HVACCs. Nb.E8 intrinsically inhibited current amplitude, sped up inactivation, and right-shifted the voltage-dependence of activation of CaVβ1-bound channels. Understanding the principles of how nb.E8 alters the biophysical properties of HVACCs may enable the rational design of small molecules that can achieve similar objectives. Interestingly, the binding site for nb.E8 is on CaVβ1 SH3 rather than the NK domain that mediates high affinity binding to α1 subunits31,32,33. Some previous efforts to develop CaV1/CaV2 channel inhibitors have focused on disrupting the association of CaVβ NK with the AID in α1 subunits using either small molecules52,53 or peptides35. Our findings suggest the possibility that small molecules that target CaVβ SH3 domains could be a novel class of effective CaV1/CaV2 channel inhibitors. Missense mutations in HVACC CaVα1 subunits that causes devastating neurological and cardiovascular diseases often do so by altering channel gating properties54,55,56,57. Thus, molecules that can correct mutation-induced gating changes in HVACCs have prospective use as therapeutics for CACNA1- and CACNA2-associated rare ion channelopathies. Our results also motivate the search for other CaVβ-targeted nanobodies that can modulate HVACC channel gating in unique ways.
The identification of a nanobody that modulates HVACCs in a CaVβ-isoform-specific manner gives reason to wonder whether there are endogenous proteins in cells that mediate similar functions, as supported by observation of a commonly targeted protein-protein interaction on the SH3 domain surface (Supplementary Fig. 3a)36. The principle of natural proteins that powerfully modulate function of HVACCs via binding to auxiliary CaVβ subunits is already well-established with RGK (Rad, Rem, Rem2, Gem) monomeric G-proteins which act as potent HVACC inhibitors6,58,59. Indeed, relief of this inhibition by PKA-mediated phosphorylation is the mechanism underlying β-adrenergic stimulation of cardiac contractility essential for the fight-or-flight response60. Beyond HVACCs, other ion channel families are also often comprised of pore-forming α1 proteins assembled with auxiliary subunits that have multiple isoforms. Thus, the principle of post-translational inhibition of channels in an auxiliary subunit-isoform-specific manner is one that is generally applicable. Our description of Chisel-1 provides a blueprint for how this critical gap in deducing the functions of multi-subunit membrane proteins can be bridged.
Methods
Nanobody generation
CaVβ1 and CaVβ3 purifications and nanobody generation were described previously61. Briefly one llama was immunized with an initial injection of 600 μg purified CaVβ1b and CaVβ3, with four boosters of 200 μg each protein administered every other week (Capralogics Inc., Hardwick, MA). 87 days after the first immunization, lymphocytes were isolated from blood sample and nanobody sequences amplified using a two-step nested PCR. The amplified Vhh genes were cloned into the phagemid plasmid, pComb3xSS (gift from Carlos Barbas; Addgene plasmid # 63890)62, and a phage display library constructed using electrocompetent TG1 E. coli cells (Lucigen). Three rounds of phage display were performed63 using 100 nM biotinylated CaVβ1b as bait on neutravidin-coated Nunc-Immuno plates (Thermo Scientific). Multiple clones of interest including nb.F3 and nb.E8 were cloned into mammalian expression systems for further characterization.
Molecular biology and plasmid construction
For flow-FRET assays, candidate CaVβ nanobodies were cloned into pCDNA3.1 and upstream of a Venus marker using EcoRI/HindIII cloning sites. CaVβ subunits were cloned into the PiggyBac CMV mammalian expression vector and downstream of a Cerulean marker using NotI/MluI cloning sites. Subsequent CaVβ cloning (SH3, NK modules, chimeras) were done using Gibson cloning64.
A customized bicistronic vector (xx-P2A-CFP) was synthesized in pUC57 vector in which coding sequence for P2A peptide was sandwiched between an upstream multiple cloning site and enhanced cyan fluorescent protein (CFP) (Genewiz). The xx-P2A-CFP fragment was amplified by PCR and cloned into the PiggyBac CMV mammalian expression vector (System Biosciences) using NheI/NotI sites. To generate nb.E8 -P2A-CFP, we PCR amplified the coding sequence for nb.E8 and cloned it into xx-P2A-CFP using NheI/AflII sites. A similar backbone was created in the PiggyBac CMV mammalian expression vector in which CFP-P2A-xx contained a multiple cloning site downstream of the P2A site (Genewiz). Nb.E8 was PCR amplified and ligated into the vector with BglII/AscI sites. The HECT domain of human Nedd4L65 (a gift from Joan Massague, Addgene plasmid # 27000) consisting of residues 596-975 was PCR amplified and inserted downstream of nb.E8 using AscI/AgeI sites. Mutagenesis of C942S was accomplished using site-directed mutagenesis.
α1B-BBS, harboring two tandem 13 residue bungarotoxin-binding sites (SWRYYESSLEPYPD) in the domain IV S5-S6 extracellular loop, was a kind gift from Dr. Steven Ikeda (NIAAA). α1C and α1C-BBS, and α1C-BBS-YFP have been described previously6,66.
Virus generation
Generation of nb.E8-IRES-mCherry and nb.E8-Nedd4L-IRES-mCherry adenoviruses was performed by Vector Biolabs (Malvern, PA).
For lentivirus construction, we followed established protocols67 using packaging plasmids kindly provided by Dr. David Baltimore. CalFectin was used to transfect the packaging plasmids VSV-G and ΔP along with the insert containing plasmid nb.E8-p2a-mCherry. Confluent HEK293T cells grown in 10 cm dishes were transfected and the media was exchanged the following morning to hippocampal culture media containing: Neurobasal media (Thermo Fisher Scientific), B-27 supplement (Thermo Fisher Scientific), and Glutamax supplement (Thermo Fisher Scientific). Media was collected approximately 48 h after transfection and centrifuged at 500 g for five minutes to pellet debris. Supernatants were collected, aliquoted, and stored at −80 °C.
HEK293 cell culture and transfection
Human embryonic kidney (HEK293) cells were a kind gift from the laboratory of Dr. Robert Kass (Columbia University). Low passage HEK293 cells were cultured at 37 °C in DMEM supplemented with 5% fetal bovine serum (FBS) and 100 mg/mL of penicillin–streptomycin. HEK293 cell transfection was accomplished using the calcium phosphate precipitation method. Briefly, plasmid DNA was mixed with 7.75 μL of 2 M CaCl2 and sterile deionized water (to a final volume of 62 μL). The mixture was added dropwise, with constant tapping to 62 μL of 2x HEPES buffered saline containing (in mM): HEPES 50, NaCl 280, Na2HPO4 1.5, pH 7.09. The resulting DNA–calcium phosphate mixture was incubated for 20 min at room temperature and then added dropwise to HEK293 cells (60–80% confluent). Cells were washed with Ca2+-free phosphate buffered saline after 4–6 h and maintained in supplemented DMEM.
Guinea pig cardiomyocyte isolation and culture
Isolation of adult guinea pig cardiomyocytes was performed in accordance with the guidelines of Columbia University Animal Care and Use Committee. Prior to isolation, plating dishes were precoated with 15 µg/mL laminin (Gibco). Adult male Hartley guinea pigs (Charles River) were euthanized with 5% isoflurane, hearts were excised and ventricular myocytes isolated by first perfusing in KH solution (mM): 118 NaCl, 4.8 KCl, 1 CaCl2 25 HEPES, 1.25 K2HPO4, 1.25 MgSO4, 11 glucose, .02 EGTA, pH 7.4, followed by KH solution without calcium using a Langendorff perfusion apparatus. Enzymatic digestion with 0.3 mg/mL Collagenase Type 4 (Worthington) with 0.08 mg/mL protease and .05% BSA was performed in KH buffer without calcium for six minutes. After digestion, 40 mL of a high K+ solution was perfused through the heart (mM): 120 potassium glutamate, 25 KCl, 10 HEPES, 1 MgCl2, and .02 EGTA, pH 7.4. Cells were subsequently dispersed in high K+ solution. Healthy rod-shaped myocytes were cultured in Medium 199 (Life Technologies) supplemented with (mM): 10 HEPES (Gibco), 1x MEM non-essential amino acids (Gibco), 2 L-glutamine (Gibco), 20 D-glucose (Sigma Aldrich), 1% vol vol−1 penicillin-streptomycin-glutamine (Fisher Scientific), 0.02 mg/mL Vitamin B-12 (Sigma Aldrich) and 5% (vol/vol) FBS (Life Technologies) to promote attachment to dishes. After 5 h, the culture medium was switched to Medium 199 with 1% (vol/vol) serum, but otherwise supplemented as described above. Cultures were maintained in humidified incubators at 37 °C and 5% CO2.
In vivo gene transfer via muscle electroporation
For in vivo electroporation experiments, animal procedures and protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Maryland. Male C57BL/6 J mice (Charles River, Wilmington, MA) were used. All mice used (9 mice) were between 30–60 days of age. Environmental conditions were maintained with a 12-h light/dark cycle and constant temperature (21–23 °C) and humidity (55 ± 10%). The cages contained corncob bedding (Harlan Teklad 7902) and environmental enrichment (cotton nestlet). Mice were supplied with dry chow (irradiated rodent diet; Harlan Teklad 2981) and water ad libitum.
Electroporation was carried out on 4-week-old C57BL mice. The intramuscular injection of various DNA plasmids was conducted, with minor modifications, according to previous reports68,69. Briefly, one footpad of an anesthetized mouse is injected subcutaneously with 20–30 μl of 3 mg/ml hyaluronidase through a 33-gauge needle. Then, 1 to 2 h later, the mouse is again anesthetized and ~40 μg of plasmid DNA is injected into the footpad. Ten minutes later, two surgical stainless-steel electrodes are placed subcutaneously close to the proximal and distal tendons of the flexor digitorum brevis (FDB) muscle and 20 pulses of 100 V/cm, 20 ms in duration, are applied at 1 Hz via a commercial high current capacity output stage (ECM 830, BTX, Harvard Apparatus, Holliston, MA). One to two weeks later, single muscle fibers are enzymatically dissociated from the injected FDB muscles and cultured as described below.
Skeletal muscle fiber culture
Culture of flexor digitorum brevis (FDB) was carried out as previously described70,71. Animals were euthanized by asphyxiation via CO2 followed by cervical dislocation according to protocols approved by the University of Maryland Institutional Animal Care and Use Committee. Briefly, the FDB muscle was isolated from male adult mice, enzymatically dissociated with collagenase type I (Sigma-Aldrich, St. Louis, MO) in MEM (Life Technologies, Carlsbad, CA) with 10% FBS, and 50 µg/ml gentamicin for 3–4 h at 37 °C. Muscle was then gently triturated to separate fibers in MEM with FBS and gentamicin. Fibers were plated in MEM culture media with 10% FBS on glass-bottomed dishes (Matek Cor. Ashland, MA, Cat. No. P35G-1.0-14-C,) coated with laminin (Thermo Fisher, Rockford, IL, Cat. No. 23017-015). Fibers were maintained in culture for 1 to 2 days at 37 °C, 5% CO2 prior to the experiments. Positively transfected fibers were identified by the CFP expression profile.
Two-electrode voltage clamp (TEVC)
The TEVC was used to measure non-linear capacitive currents and L-type Ca2+ current elicited by step depolarizations. Muscle fibers (<500 µm in length) were chosen and visualized on a Zeiss Axiovert 200 M inverted microscope. The external recording solution composition to measure non-linear capacitive currents was (in mM): 150 TEA-CH3SO3, 10 HEPES, 0.5 CaCl2, 1 MgCl2, 0.5 CdCl2 and 0.5 CoCl2, 0.001 TTX, 0.5 4-aminopiridine, 0.025 BTS (N-benzyl-p-toluene sulphonamide; Sigma-Aldrich, St Louis, MO, Cat No. 203895), pH adjusted to 7.4 with CsOH. To measure Ca2+ currents, 10 mM Ca2+ was used as charge carrier and Cd2+and Co2+ were not added to the recording solution. The current injecting electrode (V1) was filled with 2 M cesium aspartate and voltage measuring electrode (V2) was filled with 3 M cesium chloride as previously described69. Microelectrode V1 was placed at the middle of the selected fiber, and V2 was positioned halfway between the middle and the end of the selected fiber.
We used an AxoClamp 900 A and Axon Digidata 1550B low-noise digitizer (Molecular Devices, San Jose, CA, USA), HS-9A x1 (V1) and HS-9A x0.1 (V2) headstages and borosilicate glass (Warner Instruments, Cat No. G150TF-3) with resistances of 10–20 MΩ when filled with the electrode solution. Once the fibers were impaled with both microelectrodes, cells were held at −80 mV. Fibers with signs of clamp error, such as unstable holding current or rapid drifts on holding potential, were rejected from the analysis. Measurements started 3 min after TEVC clamp configuration was established. Voltage protocols were generated and current responses were digitized and stored using Clampex and Clampfit (version 11, Molecular Devices, San Jose, CA, USA). Command pulses were delivered at 30 s intervals to the levels and duration indicated in each figure from a holding potential of −80 mV, unless otherwise indicated. Currents were typically low-pass-filtered at 3–10 kHz (3-pole Bessel filter). Currents were sampled at 10 kHz. Linear capacitive and ionic currents were routinely subtracted by a P/4 protocol72. Gating charge moved during each test depolarization (QON) was quantified by calculating the area under the curve of each trace of non-linear current using the post-transient level of each trace as a steady-state value of non-linear ionic current. Total charge moved during repolarization (QOFF) was calculated similarly73,74. Total charge movement was normalized to the linear fiber capacitance, which was determined by measuring linear capacitive current elicited by a ± 5 mV test pulse from the holding potential and integrating the area under the capacitive current trace to estimate Q.
Data analysis was performed using Clampfit 8.0 (Molecular Devices, San Jose, CA, USA). Further data evaluation, non-linear fitting and statistical analysis were conducted using OriginPro 2020b software. The I–V plots from muscle fibers were fitted to a Boltzmann-Ohmic function, described by the following equation75:
where Gmax is the maximum conductance, V is the membrane potential, Vrev is the reversal potential, Vhalf is the half-activation potential, and k is a measure of the steepness.
Similarly, the Q-V relationship of each individual fiber was fitted to a single Boltzmann function, as described by the equation:
where Qmax gives the maximum charge movement, Vhalf defines the potential where Q = 0.5 of Qmax and 1/k is a measure of the steepness of the Q–V relationship.
Hippocampal isolation and culture
E18 Sprague Dawley Rat hippocampal tissue was purchased from Transnetyx Tissue Inc and disassociated immediately using 2 mg/mL papain (Worthington Biochemical Corporation). Cells were initially cultured in media containing: Neurobasal media (Thermo Fisher Scientific), B-27 supplement (Thermo Fisher Scientific), and Glutamax supplement (Thermo Fisher Scientific), and 25 μM glutamate (Sigma Aldrich). 50% of the media was exchanged every 4 days with the fresh media containing no glutamate. Cells were plated onto 12 mm #1 round coverslips coated with Poly-D lysine and laminin (Corning).
Flow cytometry-based FRET
Cells were transfected using polyethylenimine (PEI) 25 kDa linear polymer (Polysciences number 2396602). 1.5 μg of cerulean (Cer)- and venus (Ven)-tagged cDNA pairs were mixed together in 100 μl of serum-free DMEM media and 5 μl of PEI was added to each sterile tube. FRET experiments were performed two days post-transfection. The protein-synthesis inhibitor cycloheximide (100 μM) was added to cells 2 h before experimentation to halt synthesis of new fluorophores, in order to allow existing fluorophores to fully mature.
For FRET measurements, we used an LSR II (BD Biosciences) flow cytometer, equipped with 405 nm, 488 nm, and 633 nm lasers for excitation and 18 different emission channels. Forward- and side-scatter signals were detected and used to gate for single and healthy cells. To determine FRET efficiency, we measured three distinct fluorescence signals: first, SCer (corresponding to emission from the cerulean tag) is measured through the BV421 channel (excitation, 405 nm; emission, 450/50); second, SVen (corresponding to emission from the venus tag) is measured via the FITC channel (excitation, 405 nm; dichroic, 505LP; emission, 525/50); and third, SFRET (corresponding to FRET emission) is measured via the BV510 channel (excitation, 405 nm; dichroic, 505LP; emission, 525/50). These raw fluorescence measurements are subsequently used to obtain Vendirect (venus emission due to direct excitation), Cerdirect (cerulean emission due to direct excitation), and VenFRET (venus emission due to FRET excitation). Fluorescence data were exported as FCS 3.0 files for further processing and analysis using custom MATLAB (2012b) functions (MathWorks).
For each experimental run on the flow cytometer, we performed several control experiments. First, the background fluorescence level for each fluorescent channel (BGCer, BGVen and BGFRET) was obtained by measuring fluorescence from cells exposed to PEI without any fluorophore-containing plasmids. Second, cells expressing the Ven fluorophore alone were used to measure the spectral crosstalk parameter RA1, corresponding to bleed-through of Ven fluorescence into the FRET channel. Third, cells expressing the Cer fluorophore alone were used to measure spectral crosstalk parameters RD1 and RD2, corresponding to bleed-through of Cer fluorescence into the FRET and Ven channels respectively. Fourth, FRET measurements also require determination of instrument-specific calibration parameters gVen/gCer and fVen/fCer, which are respectively ratios of fluorescence excitation and emission for Ven to Cer fluorophores. These parameters also incorporate fluorophore-dependent aspects, including molar extinction (for g) and quantum yield (for f), as well as instrument-specific parameters, including laser power, attenuation by optical components, and photodetection, amplification and digitization of fluorescence. To determine these parameters, we used Cer–Ven dimers with four different linker lengths (5, 32, 50 and 228). Fifth, co-expression of Cer and Ven fluorophores provided estimates for concentration-dependent collisional FRET.
In our experiments, RA1 was approximately 0.11, RD1 approximately 2.8, and RD2 approximately 0.006. We observed only minor day-to-day variation in these measurements. For each cell, spectral cross-talk was subtracted as follows:
Following spectral unmixing, we obtained gVen/gCer and fVen/fCer from data for Cer–Ven dimers by determining the slope and intercept for the following linear relationship:
For typical experiments, gVen/gCer = 0.0194 and fVen/fCer = 2.3441. Having obtained these calibration values, we computed donor-centric FRET efficiencies as:
For Cer–Ven dimers, we obtained FRET efficiencies of roughly 0.55, 0.38, and 0.05 for linker lengths 5, 32, and 228 respectively. The relative proportion of Cer and Ven fluorophores in each cell was determined:
(8) NCer = Cerdirect/(1 – ED)
(9) NVen = Vendirect/(gVen/gCer × fVen/fCer). To construct FRET two-hybrid-binding curves, we imposed a 1:1 binding isotherm as in previous studies29,76. For each FRET pair, we obtained effective dissociation constant (Kd,EFF), ED,max and 95% confidence intervals by constrained least-squares fit.
Flow cytometry assay of total and surface calcium channels
Cell surface and total ion channel pools were assayed by flow cytometry in live, transfected HEK293 cells as previously described77. Briefly, 48 h post-transfection, cells cultured in 12-well plates gently washed with ice cold PBS containing Ca2+ and Mg2+ (in mM: 0.9 CaCl2, 0.49 MgCl2, pH 7.4), and then incubated for 30 min in blocking medium (DMEM with 3% BSA) at 4 °C. HEK293 cells were then incubated with 1 μM Alexa Fluor 647 conjugated α-bungarotoxin (BTX647; Life Technologies) in DMEM/3% BSA on a rocker at 4 °C for 1 h, followed by washing three times with PBS (containing Ca2+ and Mg2+). Cells were gently harvested in Ca2+-free PBS, and assayed by flow cytometry using a BD Fortessa Cell Analyzer (BD Biosciences, San Jose, CA, USA) running BD FACSDiva (v8.65) acquisition software. CFP- and YFP-tagged proteins were excited at 407 and 488 nm, respectively, and Alexa Fluor 647 was excited at 633 nm.
Electrophysiology
Whole-cell recordings of HEK293 cells were conducted 48 h after transfection using an EPC-10 patch clamp amplifier (HEKA Electronics) controlled by pulse software (HEKA Pulse v8.65). Micropipettes were prepared form 1.5 mm thin-walled glass (World Precision Instruments) using a P97 microelectrode puller (Sutter Instruments). Internal solution contained (mM): 135 cesium-methanesulfonate (CsMeSO3), 5 CsCl, 5 EGTA, 1 MgCl2, 2 MgATP, and 10 HEPES (pH 7.3). Series resistance was typically between 1–2 MΩ. There was no electronic resistance compensation. External solution contained (mM): 140 tetraethylammonium-MeSO3, 5 BaCl2, and 10 HEPES (pH 7.4). Whole-cell I-V curves were generated from a family of step depolarizations (−60 mV to +80 mV from a holding potential of −90 mV). Currents were sampled at 20 kHz and filtered at 5 kHz. Traces were acquired at a repetition interval of 10 s. Leak and capacitive transients were subtracted using a P/4 protocol.
Cell-attached single-channel recordings were performed at room temperature as described37. Patch pipettes (5–10 MΩ) were pulled from ultra-thick-walled borosilicate glass (BF200-116-10, Sutter Instruments), and coated with Sylgard. Currents were filtered at 2 kHz. The pipette solution contained 140 mM tetraethylammonium methanesulfonate; 10 mM HEPES; 40 mM BaCl2; at 300 mOsm l−1, adjusted with tetraethylammonium methanesulfonate; and pH 7.4, adjusted with tetraethylammonium hydroxide. To maintain the membrane potential at 0 mV, the bath contained 132 mM potassium glutamate, 5 mM KCl, 5 mM NaCl, 3 mM MgCl2, 2 mM EGTA, 10 mM glucose, 20 mM HEPES; at 300 mOsm L−1, adjusted with glucose; and pH 7.4, adjusted with sodium hydroxide. Cell-attached single-channel currents were measured during 200 ms voltage ramps between −80 mV and +70 mV (portions between −50 mV and +40 mV are displayed and analyzed). For each patch, we recorded 80–120 sweeps with a repetition interval of 10 s.
Whole-cell recordings of guinea pig cardiomyocytes were performed 48 h after infection, with internal solution comprised of (mM): 150 CsMeSO3, 10 EGTA, 5 CsCl, MgCl2, 4 MgATP, and 10 HEPES. For formation of gigaohm seals and initial break-in to the whole-cell configuration, cells were perfused in Tyrode solution containing (mM): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 0.33 NaH2PO4, and 10 HEPES (pH 7.4). Upon successful break-in, the perfusing media was switched to an external solution composed of (mM): 155 N-methyl-D-glucamine, 10 4-amino-pyridine, 1 MgCl2, 5 BaCl2, and 10 HEPES (pH 7.4). Currents were sampled at 20 kHz and filtered at 5 kHz. Leak and capacitive transients were subtracted using a P/4 protocol.
Immunofluorescence staining
Approximately 48 h after adenoviral infection guinea pig cardiomyocytes were fixed in 4% paraformaldehyde (wt/vol, in PBS) for 20 min at RT. Cells were washed twice with PBS and then incubated in 0.1 M glycine (in PBS) for 10 min at RT to block free aldehyde groups. Fixed cells were then permeabilized with 0.2% Triton X-100 (in PBS) for 20 min at RT. Non-specific binding was blocked with a 1 h incubation at RT in PBS solution containing 3% (vol vol−1) normal goat serum (NGS), 1% BSA, and 0.1% Triton X-100. Cells were then incubated with rabbit anti-CaV1.2 primary antibody (Alomone Labs, 1:1000) in PBS containing 1% NGS, 1% BSA, and 0.1% BSA overnight at 4 °C. Cells were washed three times for 10 min each with PBS containing 0.1% Triton X-100 and then stained with anti-rabbit 488 secondary antibody (Thermofisher, 1:1000) for 1 h at RT. Antibody dilutions were prepared in PBS solution containing 1% NGS, 1% BSA, and 0.1% Triton X-100. The cells were then washed in PBS with 0.1% Triton X-100 and imaged in the same solution.
Hippocampal neurons were fixed with ice-cold 4% paraformaldehyde in phosphate buffered saline supplemented with 4% sucrose at room temperature for 10 min. Cells were then washed 3x in PBS, permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature, and washed 3x in PBS. The cells were blocked at room temperature for 1 h in PBS + 1% BSA + 3% normal goat serum + 0.1% Triton X-100, and then incubated in primary antibody, diluted in PBS + 0.1% Triton X-100 + 1% BSA + 3% NGS overnight. Primary antibodies: anti-pCREB (1:333 dilution, Cell Signaling Technology), MAP2 (1:1000 dilution, Santa Cruz Biotechnology), CaVβ1 (1:200 dilution, Alomone). The following day, cells were washed 4x with PBS, then incubated in secondary antibody diluted in PBS + 0.1% Triton X-100 + 1% BSA + 3% NGS. Secondary antibodies: anti-rabbit Alexa-488, anti-mouse Alexa-647 (all 1:1000, Thermo Fisher Scientific). Hippocampal neurons were imaged using Nikon Eclipse Ti A1-A laser scanning confocal microscope with NIS-Elements AR 5.02.00 64-bit software.
Pulldown assays
60 mm dishes of transfected HEK293 cells were harvested in PBS, centrifuged at 2000 g (4 °C) for 5 min and the pellet resuspended in NP40 lysis buffer containing (mM): 150 NaCl, 50 Tris (pH 8), 1% Triton X-100, and supplemented with protease inhibitor mixture (10 μL mL−1, Sigma Aldrich), 1 PMSF, 2 N-ethylmaleimide, .05 PR-619 deubiquitinase inhibitor (LifeSensors). Cells were lysed on ice for 1 h with intermittent vortexing and centrifuged at 10,000 g for 10 min (4 °C). The soluble lysate was collected and protein concentration determined with the bis-cinchonic acid protein estimation kit (Pierce Technologies).
For CaVβ1 pulldowns, lysates were precleared with 10 μL of protein A/G sepharose beads (Rockland) for 1 h at 4 °C and then incubated with 2 µg anti-CaVβ1 antibody (UC Davis/NIH NeuroMab Facility, clone N7/18) for 1 h at 4 °C. Equivalent amounts of protein were then added to spin columns with 25 μL equilibrated protein A/G Sepharose beads and rotated overnight at 4 °C. Immunoprecipitates were washed a total of five times with NP40 buffer and then eluted with 30 μL elution buffer (50 mM Tris, 10% (vol vol−1) glycerol, 2% SDS, 100 mM DTT, and 0.2 mg mL−1 bromophenol blue) at 55 °C for 15 min. For CaV1.2 α1C pulldowns, lysates were added to spin columns containing 10 μL of equilibrated RFP-trap agarose beads, rotated at 4 °C for 1 h, and then washed/eluted as described above. Proteins were resolved on a 4–12% Bis Tris gradient precast gel (Life Technologies) in MOPS-SDS running buffer (Life Technologies) at 200 V constant for ~1 h. Protein bands were transferred by tank transfer onto a polyvinylidene difluoride (PVDF, EMD Millipore) membrane in transfer buffer (25 mM Tris pH 8.3, 192 mM glycine, 15% (vol/vol) methanol, and 0.1% SDS). The membranes were blocked with a solution of 5% nonfat milk (BioRad) in tris-buffered saline-tween (TBS-T) (25 mM Tris pH 7.4, 150 mM NaCl, and 0.1% Tween-20) for 1 h at RT and then incubated overnight at 4 °C with primary antibodies (anti-FLAG HRP, Sigma Aldrich; Actin, Sigma Aldrich; α1C, Alomone) in blocking solution. The blots were washed with TBS-T three times for 10 min each and then incubated with secondary horseradish peroxidase-conjugated antibody for 1 h at RT. After washing in TBS-T, the blots were developed with a chemiluminescent detection kit (Pierce Technologies) and then visualized on a gel imager. Membranes were then stripped with harsh stripping buffer (2% SDS, 62 mM Tris pH 6.8, 0.8% β-mercaptoethanol) at 50 °C for 30 min, rinsed under running water for 2 min, and washed with TBST (3x, 10 min). Membranes were pre-treated with 0.5% glutaraldehyde and re-blotted with anti-ubiquitin (VU1, LifeSensors, 1:500) as per the manufacturers’ instructions.
Western blot
Hippocampal cultures were harvested in PBS, centrifuged at 500 g for 5 min, then resuspended in NP40 lysis buffer (as above). 25 μg of protein per sample was loaded onto a PVDF membrane and probed as above using Cavβ1, Cavβ2, Cavβ3 (Alomone, 1:1000), Cavβ4 (NeuroMab, 1:1000) and actin (Sigma Aldrich, 1:1000).
Skeletal muscle Ca2+ imaging
Fiber loading with rhod-2 AM, a membrane-permeable non-ratiometric high affinity Ca2+ indicator (Thermo Fisher, Cat. No. R1244), and subsequent imaging and analyses were performed as previously described78,79 but with some modifications. Briefly, cultured FDB fibers were loaded with rhod-2 (2 μM for 60 min at 22 C) in 1 mL of L-15 media (ionic composition in mM: 137 NaCl, 5.7 KCl, 1.26 CaCl2, 1.8 MgCl2, pH 7.4; Life Technologies, Carlsbad, CA) supplemented with 0.25% w/v bovine serum albumin (BSA; Sigma‐Aldrich, St Louis MO, Cat. No. A‐7906). The fibers were washed thoroughly with appropriate L-15 media to remove residual fluorescent dye. All single fiber recordings were performed at room temperature. Confocal imaging of rhod-2 (100 µs/line) was performed using high-speed confocal system LSM 5 Live system (Carl Zeiss, Jena, Germany). Rhod-2 was excited with a 532 nm laser, and the fluorescence emitted >550 nm was detected on a Zeiss Axiovert 200 M inverted microscope and confocal imaging was performed in line scan xt mode as previously described80, with images acquired for 0.4 to 1 s, using a 63 × 1.2 N.A. water immersion objective.
Hippocampal calcium imaging
Hippocampal neurons were washed twice in basal solution containing (mM): 150 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 D-glucose, pH 7.4, and incubated in the same solution containing 2 μM fura-2 with 0.05% Pluronic F-127 detergent (Life Technologies) for 30 min at 37 °C, 5% CO2. Cells were subsequently washed twice in the same solution and placed on an inverted Nikon Ti-eclipse microscope with a Nikon Plan fluor 20x objective (0.45 N.A.). Fura-2 measurements were recorded at excitation wavelengths of 340 and 380 nm using EasyRatioPro (HORIBA Scientific). Hippocampal neurons were depolarized with a solution in which NaCl was reduced to 65 mM and KCl increased to 90 mM.
Excitation-transcription coupling
Cultured hippocampal neurons were infected with lentivirus approximately 7 days after plating and used for experiments 7–10 days afterwards. Experiments were performed as previously described81. Cells were pre-incubated for 4 h in culture media supplemented with: 10 μM NBQX (Tocris Biosciences), 10 μM AP5 (Tocris Biosciences), and 0.5 μM tetrodotoxin (Tocris Biosciences). All solutions used throughout the experiment contained these blockers. Cells were then washed twice in hippocampal basal imaging solution (as above), and incubated for 5 min in 5 K basal solution (+ any calcium channel blockers). Cells were then placed in 90 K solution (plus indicated calcium channel blockers) for 3 min and immediately fixed with ice-cold 4% paraformaldehyde in phosphate buffered saline supplemented with 4% sucrose. Samples were immediately processed for immunostaining, as described above.
Protein expression and purification
Cavβ2a-link34, a construct of rat Cavβ2a (GenBank NM_053851) consisting of the SH3 domain (residues 17–138) and the NK domain (residues 203–425) separated by a serine residue34, a construct covering rat Cavβ1b (GenBank NM_059042.2) residues 58 to 427, nb.F3, and nb.E8 were expressed using pET28HMT31, a vector containing in series a hexahistidine tag, maltose binding protein (MBP), and Tobacco Etch Virus (TEV) protease site N-terminal to the protein of interest. Cavβ1b nb.F3, and nb.E8 were cloned into this vector by Gibson assembly64.
Cavβ2a-Link was expressed and purified as described previously34. For Cavβ1b−58-427, 100 ng of plasmid was transformed into RosettaTM(DE3)pLysS competent cells, plated on LB (Luria Broth) agar containing 50 μg ml−1 Kanamycin and 34 μg ml−1 Chloramphenicol, and grown overnight at 37 °C. A single colony was used to inoculate 60 ml LB media containing 50 μg ml−1 Kanamycin and 34 μg ml−1 Chloramphenicol and was grown overnight at 37 °C. Six 1 L flasks containing 2x YT media (5 g NaCl, 16 g tryptone, 10 g yeast extract) supplemented with 50 μg ml−1 Kanamycin and 34 μg ml−1 Chloramphenicol were each inoculated with 10 ml of overnight starter culture per liter and grown at 37 °C in a shaker incubator until OD600 reached 0.4, at which point the temperature was changed to 18 °C and cultures were induced with 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) after 30 minutes and then grown overnight. Cells were harvested by centrifugation (4000 g × 20 min). Cell pellets were flash frozen in liquid nitrogen and stored at −80 °C until further use. All protein purification steps, unless mentioned otherwise, were carried out at 4 °C. 6 L worth of bacterial pellets were suspended in 100 ml of ice-cold lysis buffer containing 200 mM KCl, 20 mM Imidazole, 10% glycerol, 1 mM Phenylmethylsulphonyl fluoride (PMSF), 0.1 mg ml−1 DNaseI, 10 mM HEPES/KOH, pH 7.4 and lysed using an EmulsiFlex-C5 homogenizer (Avestin) (2 passages, 5000 psi). Lysate was clarified by ultracentrifugation at 100,000 g for 1 hour at 4 °C. Supernatant was applied to a 20 ml Poros 20 MC Ni2+ column (Applied Biosystems) and equilibrated with Buffer A (200 mM KCl, 20 mM Imidazole, 10% glycerol, 10 mM HEPES/KOH pH 7.4). The column was washed with two column volumes (CVs) of Buffer A, followed by two CVs of 94% Buffer A and 6% Buffer B (200 mM KCl, 500 mM Imidazole, 10% glycerol, 10 mM HEPES/KOH pH 7.4). Bound protein was eluted using two CVs of 40% Buffer A and 60% Buffer B. Eluted protein was concentrated to <1 ml using centrifugal concentrator (Amicon filter, MWCO 50 kDa) and then diluted to 50 ml in TEV digestion buffer (200 mM KCl, 10 mM β-Mercaptoethanol (BME) 10 mM HEPES/KOH pH 7.4). Affinity tag (Histidine tagged MBP) was removed by addition of 50 μl of 1 mg ml−1 6x histidine tagged superTEV82, rotating at room temperature overnight. Cleaved protein was separated from the affinity tag and protease by passing it through POROS 20MC Ni2+ column (Applied Biosystems), equilibrated with Buffer A. Flowthrough was collected, concentrated to <1 ml using a centrifugal concentrator (Amicon filter, MWCO 30 kDa), diluted to 50 ml in buffer B50 (50 mM KCl, 20 mM HEPES/KOH pH 7.4, 10 mM BME), and applied to a 10 ml Hiload SP HP column (Cytiva Life Sciences) for ion exchange chromatography. Protein was eluted using a linear gradient of 0-100% buffer B1000 (1000 mM KCl, 20 mM HEPES/KOH pH 7.4, 10 mM BME) over 20 CVs. Purity was validated using SDS-PAGE. Fractions containing highest purity were pooled and concentrated to >30 mg ml−1 using a centrifugal concentrator (Amicon filter, MWCO 30 kDa), flash frozen in liquid nitrogen, and stored at −80 °C.
Nb.F3 and nb.E8 were expressed and purified as follows: 100 ng plasmid was transformed into ShuffleTM competent cells and plated on LB agar plates containing 50 μl ml−1 Kanamycin and overnight a 30 °C. 60 ml LB containing 50 μl ml−1 Kanamycin was inoculated using a single colony and grown overnight at 30 °C. Six 1 L flasks of 2x YT media containing 50 μl ml−1 Kanamycin were each inoculated with 10 ml of starter culture and grown at 30 °C until OD600 reached 0.4. Temperature of the shaker was then reduced to 18 °C and cultures were induced at 0.6 OD600 with 1 mM IPTG and grown overnight. Cells were harvested by centrifugation at 4000 g for 20 min. Bacterial pellets were flash frozen in liquid nitrogen and stored at −80 °C until further use. All protein purification steps, unless mentioned otherwise, were carried out at 4 °C. 6 L bacterial pellet was resuspended in 100 ml ice-cold lysis buffer. Resuspended cells were lysed using emulsiflex (2 passages, 5000 psi) and clarified by ultracentrifugation at 100,000 g for 1 h at 4 °C. Supernatant was applied to 20 ml Poros 20 MC Ni2+ column equilibrated with Buffer A. Column was washed with 2 CV Buffer A followed by 2 CV of 94% Buffer A and 6% Buffer B before elution with 2 CV of 40% Buffer A and 60% Buffer B. 10 ml fractions were collected and protein was concentrated to <1 ml using centrifugal concentrator (Amicon filter, MWCO 30 kDa). Eluted protein was concentrated to <1 ml using a centrifugal concentrator (Amicon filter, MWCO 10 kDa) and then diluted to 50 ml in TEV digestion buffer (200 mM KCl, 10 mM HEPES/KOH pH 7.4). Affinity tag (Histidine tagged MBP) was removed by addition of 50 μl of 1 mg ml−1 6x histidine tagged superTEV82, rotating at room temperature overnight. Cleaved protein was separated from the affinity tag and protease by passing it through POROS20MC Ni2+ column (Applied Biosystems), equilibrated with Buffer A. Flowthrough was collected, concentrated to <1 ml using centrifugal concentrator (Amicon filter, MWCO 10 kDa), and purified on a 24 ml Superdex75 10/300 GL (GE Healthcare) column equilibrated with SEC buffer (150 mM KCl, 10% Glycerol, 10 mM HEPES/KOH pH 7.4). Purity was validated using SDS-PAGE. Fractions with highest protein purity were pooled and concentrated to >30 mg ml−1 using a centrifugal concentrator (Amicon filter, MWCO 10 kDa), flash frozen in liquid nitrogen, and stored at −80 °C.
Protein crystallization and structure determination
For Cavβ2a-Link:nbF3 complex formation, 50 nmoles of Cavβ2a-Link (Molecular weight (MW)− 39.4 kDa) was incubated with 100 nmoles of nbF3 (MW- 13.78 kDa) (1:2 molar ratio) in SEC buffer with final reaction volume of 500 μl for 30 min at 4 °C. For Cavβ1b−58-427:nbF3:nbE8 complex formation, 50 nmoles of Cavβ1b−58-427 (MW- 41.24 kDa) was combined with 100 nmoles of nb.F3 and 100 nmoles of nb.E8 (MW- 14.3 kDa) in 1:2:2 molar ratio, in total SEC buffer with final 500 μl reaction volume. The proteins were incubated for 30 min at 4 °C. Nanobody complexes were isolated by injecting the incubated protein mixes onto a 24 ml Superdex200 10/300 GL column (Cytiva Life Sciences) equilibrated with SEC buffer. Complex formation was validated by SDS PAGE, concentrated to ~10 mg ml−1 using centrifugal concentrator (Amicon filter, MWCO 3 kDa), and used immediately for crystallization by sitting drop vapor diffusion at 4 °C using 1:1 ratios of protein:precipitant. Protein concentrations were determined by absorbance at 280 nm83.
The Cavβ2a-Link:nbF3 complex crystallized in 30–40% MPD, 0.1 M HEPES/KOH pH 7.0. Crystals were harvested at 4 °C using 40% MPD as cryoprotectant. The Cavβ1b−58-427:nb.F3:nb.E8 complex crystallized in 15–22% PEG3350, 0.15–0.25 M Ammonium Sulphate. Crystals were cryoprotected by gradual increment of glycerol concentration, starting from 5% to 30%, in mother liquor. Finally, the crystals were harvested with 30% glycerol as cryoprotectant.
Diffraction data were collected at 100 K at Beamline 8.3.1 (Advanced Light Source, Lawrence Berkeley National laboratories) and APS GM/CAT beamline 23-IDB/D Chicago, Illinois. Data was indexed using XDS84 and scaled and merged using AIMLESS85. Molecular replacement was done in PHASER86 using PDB:5V2P chain A35. Model was built and improved by successive rounds of building and refinement using COOT87 and Phenix88, respectively.
Data and statistical analysis
Data were analyzed off-line using FloJo, PulseFit, Microsoft Excel, Origin and GraphPad Prism software. Statistical analyses were performed in Origin, Microsoft Excel or GraphPad Prism using built-in functions. Statistically significant differences between means (P < 0.05) were determined using Student’s t test for comparisons between two groups or one-way ANOVA for multiple groups, with Tukey’s post-hoc analysis. Data are presented as means ± SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon reasonable request. Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession codes 8DAM (CaVβ1b:nb.E8) and 8E0E (CaVβ2a:nb.F3). Previously published PDB can be accessed via accession code 5V2P [https://doi.org/10.2210/pdb7VF9/pdb] (CaVβ2a:CaV1.2 AID peptide complex). The source data underlying Fig. 2a, b, i, Fig. 4b, c, e, f, h, i, k, l, Fig. 5c, g, Fig. 6c-f, h-k, Fig. 7a–c, Fig. 8c, d, g, h, Fig. 9b, d, f, Supplementary Fig. 5, Supplementary Fig. 7b, and Supplementary Fig. 8b are provided as a Source Data File. Source data are provided with this paper.
Code availability
Flow-FRET and single channel recordings were analyzed using custom Matlab (2012b) software. The code is accessible at: https://github.com/manubenjohny/FACS_FRET and https://github.com/manubenjohny/SinglesAnalysis/, respectively.
References
Hille, B. Ion Channels of Excitable Membranes. Third edn, (Sinauer Associates, Inc., 2001).
Zamponi, G. W., Striessnig, J., Koschak, A. & Dolphin, A. C. The Physiology, Pathology, and Pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharm. Rev. 67, 821–870 (2015).
Buraei, Z. & Yang, J. The {beta} subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506 (2010).
Jones, L. P., Wei, S. K. & Yue, D. T. Mechanism of auxiliary subunit modulation of neuronal alpha1E calcium channels. J. Gen. Physiol. 112, 125–143 (1998).
Altier, C. et al. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J. Biol. Chem. 277, 33598–33603 (2002).
Yang, T., Xu, X., Kernan, T., Wu, V. & Colecraft, H. M. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J. Physiol. 588, 1665–1681 (2010).
Cassidy, J. S., Ferron, L., Kadurin, I., Pratt, W. S. & Dolphin, A. C. Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary alpha2delta-1 subunits. Proc. Natl Acad. Sci. USA 111, 8979–8984 (2014).
Fang, K. & Colecraft, H. M. Mechanism of auxiliary beta-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J. Physiol. 589, 4437–4455 (2011).
Altier, C. et al. The Cavbeta subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 14, 173–180 (2011).
Waithe, D., Ferron, L., Page, K. M., Chaggar, K. & Dolphin, A. C. Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradation. J. Biol. Chem. 286, 9598–9611 (2011).
Neely, A. & Hidalgo, P. Structure-function of proteins interacting with the alpha1 pore-forming subunit of high-voltage-activated calcium channels. Front. Physiol. 5, 209 (2014).
Papa, A. et al. Adrenergic CaV1.2 activation via Rad phosphorylation converges at alpha1C I-II Loop. Circ. Res. 128, 76–88 (2021).
Takahashi, S. X., Miriyala, J. & Colecraft, H. M. Membrane-associated guanylate kinase-like properties of beta-subunits required for modulation of voltage-dependent Ca2+ channels. Proc. Natl Acad. Sci. USA 101, 7193–7198 (2004).
Herzig, S. et al. Mechanism of Ca(v)1.2 channel modulation by the amino terminus of cardiac beta2-subunits. FASEB J. 21, 1527–1538 (2007).
Restituito, S. et al. The [beta]2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J. Neurosci. 20, 9046–9052 (2000).
Takahashi, S. X., Mittman, S. & Colecraft, H. M. Distinctive modulatory effects of five human auxiliary beta 2 subunit splice variants on L-type calcium channel gating. Biophysical J. 84, 3007–3021 (2003).
Zamponi, G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Disco. 15, 19–34 (2016).
Hockerman, G. H., Peterson, B. Z., Johnson, B. D. & Catterall, W. A. Molecular determinants of drug binding and action on L-type calcium channels. Annu Rev. Pharm. Toxicol. 37, 361–396 (1997).
Olivera, B. M., Miljanich, G. P., Ramachandran, J. & Adams, M. E. Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu Rev. Biochem. 63, 823–867 (1994).
Striessnig, J. et al. Structural basis of drug binding to L Ca2+ channels. Trends Pharm. Sci. 19, 108–115 (1998).
Gregg, R. G. et al. Absence of the beta subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the alpha 1 subunit and eliminates excitation-contraction coupling. Proc. Natl Acad. Sci. USA 93, 13961–13966 (1996).
Ball, S. L. et al. Role of the beta(2) subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol. Vis. Sci. 43, 1595–1603 (2002).
Weissgerber, P. et al. Reduced cardiac L-type Ca2+ current in Ca(V)beta2-/- embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ. Res. 99, 749–757 (2006).
Neef, J. et al. The Ca2+ channel subunit beta2 regulates Ca2+ channel abundance and function in inner hair cells and is required for hearing. J. Neurosci. 29, 10730–10740 (2009).
Murakami, M. et al. Pain perception in mice lacking the beta3 subunit of voltage-activated calcium channels. J. Biol. Chem. 277, 40342–40351 (2002).
Meissner, M. et al. Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J. Biol. Chem. 286, 15875–15882 (2011).
Yang, L. et al. Cardiac CaV1.2 channels require beta subunits for beta-adrenergic-mediated modulation but not trafficking. J. Clin. Invest 129, 647–658 (2019).
Morgenstern, T. J., Park, J., Fan, Q. R. & Colecraft, H. M. A potent voltage-gated calcium channel inhibitor engineered from a nanobody targeted to auxiliary CaVbeta subunits. eLife 8, https://doi.org/10.7554/eLife.49253 (2019).
Lee, S. R., Sang, L. & Yue, D. T. Uncovering Aberrant mutant PKA function with flow cytometric FRET. Cell Rep. 14, 3019–3029 (2016).
Kanner, S. A., Shuja, Z., Choudhury, P., Jain, A. & Colecraft, H. M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 17, 1245–1253 (2020).
Van Petegem, F., Clark, K. A., Chatelain, F. C. & Minor, D. L. Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675 (2004).
Chen, Y. H. et al. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680 (2004).
Opatowsky, Y., Chen, C. C., Campbell, K. P. & Hirsch, J. A. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399 (2004).
Van Petegem, F., Duderstadt, K. E., Clark, K. A., Wang, M. & Minor, D. L. Jr. Alanine-scanning mutagenesis defines a conserved energetic hotspot in the CaValpha1 AID-CaVbeta interaction site that is critical for channel modulation. Structure 16, 280–294 (2008).
Findeisen, F. et al. Stapled voltage-gated calcium channel (CaV) alpha-Interaction Domain (AID) peptides act as selective protein-protein interaction inhibitors of CaV function. ACS Chem. Neurosci. 8, 1313–1326 (2017).
Van Petegem, F. & Minor, D. L. Jr The structural biology of voltage-gated calcium channel function and regulation. Biochem. Soc. Trans. 34, 887–893 (2006).
Adams, P. J., Ben-Johny, M., Dick, I. E., Inoue, T. & Yue, D. T. Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell 159, 608–622 (2014). PMID - 25417111.
Colecraft, H. M. et al. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J. Physiol. 541, 435–452 (2002).
Bannister, R. A. & Beam, K. G. Ca(V)1.1: The atypical prototypical voltage-gated Ca(2)(+) channel. Biochim. Biophys. Acta 1828, 1587–1597 (2013).
Aisenberg, W. H. et al. Multiubiquitination of TRPV4 reduces channel activity independent of surface localization. J. Biol. Chem. 298, 101826 (2022).
Elliott, E. M., Malouf, A. T. & Catterall, W. A. Role of calcium channel subtypes in calcium transients in hippocampal CA3 neurons. J. Neurosci. 15, 6433–6444 (1995).
Simms, B. A. & Zamponi, G. W. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45 (2014).
Wheeler, D. G. et al. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149, 1112–1124 (2012).
Obermair, G. J. et al. Reciprocal interactions regulate targeting of calcium channel beta subunits and membrane expression of alpha1 subunits in cultured hippocampal neurons. J. Biol. Chem. 285, 5776–5791 (2010).
Pichler, M. et al. Beta subunit heterogeneity in neuronal L-type Ca2+ channels. J. Biol. Chem. 272, 13877–13882 (1997).
McEnery, M. W., Copeland, T. D. & Vance, C. L. Altered expression and assembly of N-type calcium channel alpha1B and beta subunits in epileptic lethargic (lh/lh) mouse. J. Biol. Chem. 273, 21435–21438 (1998).
Deisseroth, K., Heist, E. K. & Tsien, R. W. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392, 198–202 (1998).
Schredelseker, J. et al. The beta 1a subunit is essential for the assembly of dihydropyridine-receptor arrays in skeletal muscle. Proc. Natl Acad. Sci. USA 102, 17219–17224 (2005).
Schredelseker, J., Dayal, A., Schwerte, T., Franzini-Armstrong, C. & Grabner, M. Proper restoration of excitation-contraction coupling in the dihydropyridine receptor beta1-null zebrafish relaxed is an exclusive function of the beta1a subunit. J. Biol. Chem. 284, 1242–1251 (2009).
Jeon, D. et al. Ablation of Ca2+ channel beta3 subunit leads to enhanced N-methyl-D-aspartate receptor-dependent long term potentiation and improved long term memory. J. Biol. Chem. 283, 12093–12101 (2008).
Murakami, M. et al. Modified behavioral characteristics following ablation of the voltage-dependent calcium channel beta3 subunit. Brain Res. 1160, 102–112 (2007).
Khanna, R. et al. Targeting the CaValpha-CaVbeta interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain https://doi.org/10.1097/j.pain.0000000000001524 (2019).
Chen, X. et al. Small-molecule CaValpha1CaVbeta antagonist suppresses neuronal voltage-gated calcium-channel trafficking. Proc. Natl Acad. Sci. USA 115, E10566–E10575 (2018).
Splawski, I. et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004).
Zhang, Q., Chen, J., Qin, Y., Wang, J. & Zhou, L. Mutations in voltage-gated L-type calcium channel: implications in cardiac arrhythmia. Channels (Austin) 12, 201–218 (2018).
Striessnig, J., Bolz, H. J. & Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflug. Arch. 460, 361–374 (2010).
Pietrobon, D. CaV2.1 channelopathies. Pflug. Arch. 460, 375–393 (2010).
Beguin, P. et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411, 701–706 (2001). 35079621 [pii].
Yang, T. & Colecraft, H. M. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta 1828, 1644–1654 (2013).
Liu, G. et al. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700 (2020).
Morgenstern, T. J., Park, J., Fan, Q. R. & Colecraft, H. M. A potent voltage-gated calcium channel inhibitor engineered from a nanobody targeted to auxiliary Ca(V)β subunits. Elife 8, https://doi.org/10.7554/eLife.49253 (2019).
Andris-Widhopf, J., Rader, C., Steinberger, P., Fuller, R. & Barbas, C. F. 3rd Methods for the generation of chicken monoclonal antibody fragments by phage display. J. Immunol. Methods 242, 159–181 (2000).
Pardon, E. et al. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9, 674–693 (2014).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, nmeth.1318.
Gao, S. et al. Ubiquitin Ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol. Cell 36, 457–468 (2009).
Kanner, S. A., Morgenstern, T. & Colecraft, H. M. Sculpting ion channel functional expression with engineered ubiquitin ligases. eLife 6, e29744 (2017).
Lois, C., Hong, E. J., Pease, S., Brown, E. J. & Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 (2002).
DiFranco, M., Quinonez, M., Capote, J. & Vergara, J. DNA transfection of mammalian skeletal muscles using in vivo electroporation. J. Vis. Exp. https://doi.org/10.3791/1520 (2009).
Banks, Q. et al. Voltage sensor movements of CaV1.1 during an action potential in skeletal muscle fibers. Proc. Natl Acad. Sci. U S A 118, https://doi.org/10.1073/pnas.2026116118 (2021).
Hernández-Ochoa, E. O. et al. Elevated extracellular glucose and uncontrolled type 1 diabetes enhance NFAT5 signaling and disrupt the transverse tubular network in mouse skeletal muscle. Exp. Biol. Med. (Maywood) 237, 1068–1083 (2012).
Liu, Y., Carroll, S. L., Klein, M. G. & Schneider, M. F. Calcium transients and calcium homeostasis in adult mouse fast-twitch skeletal muscle fibers in culture. Am. J. Physiol. 272, C1919–C1927 (1997).
Armstrong, C. M. & Bezanilla, F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J. Gen. Physiol. 63, 533–552 (1974).
Prosser, B. L., Hernández-Ochoa, E. O., Zimmer, D. B. & Schneider, M. F. The Qgamma component of intra-membrane charge movement is present in mammalian muscle fibres, but suppressed in the absence of S100A1. J. Physiol. 587, 4523–4541 (2009).
Schneider, M. F. & Chandler, W. K. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature 242, 244–246 (1973).
Nakai, J. et al. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75 (1996).
Rivas, S., Hanif, K., Chakouri, N. & Ben-Johny, M. Probing ion channel macromolecular interactions using fluorescence resonance energy transfer. Methods Enzymol. (2021).
Morgenstern, T. J. & Colecraft, H. M. Controlling ion channel trafficking by targeted ubiquitination and deubiquitination. Methods Enzymol. 654, 139–167 (2021).
Robison, P. et al. Impaired calcium signaling in muscle fibers from intercostal and foot skeletal muscle in a cigarette smoke-induced mouse model of COPD. Muscle Nerve 56, 282–291 (2017).
Prosser, B. L. et al. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 283, 5046–5057 (2008).
Hernández-Ochoa, E. O., Schachter, T. N. & Schneider, M. F. Elevated nuclear Foxo1 suppresses excitability of skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 305, C643–C653 (2013).
Wheeler, Damian, G. et al. CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell 149, 1112–1124 (2012).
Blommel, P. G. & Fox, B. G. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr. Purif. 55, 53–68 (2007).
Edelhoch, H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6, 1948–1954 (1967).
Kabsch, W. Xds. Acta Crystallogr D. Biol. Crystallogr 66, 125–132 (2010).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr D. Biol. Crystallogr 69, 1204–1214 (2013).
McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D. Biol. Crystallogr 63, 32–41 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Acknowledgements
We thank Ming Chen for technical support. This work was supported by grants RO1-HL121253 and RO1-HL122421 from the NIH (to HMC), F31 DK118866 (to T.J.M.), R01-HL080050 (to D.L.M.), and R01-AR075726 and R01-NS103777 to (M.F.S.). Flow cytometry experiments were performed in the CCTI Flow Cytometry Core, supported in part by the NIH (S10RR027050). Confocal images were collected in the HICCC Confocal and Specialized Microscopy Shared Resource, supported by NIH (P30 CA013696). This work was prepared while R.A.B. was employed at the University of Maryland Baltimore. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services or the United States government.
Author information
Authors and Affiliations
Contributions
T.J.M. and H.M.C. conceived the study; T.J.M., N.N., E.H.-O., M.B.J., R.A.B., M.F.S., D.L.M., H.M.C. designed the experiments; T.J.M., E.H.-O., and J.D.L. performed electrophysiology and Ca2+ imaging experiments; N.N. and D.L.M. performed and analyzed structural biology experiments; P.C. and T.J.M. performed FLOW-fret experiments; M.B.J. analyzed flow-fret and single-channel analyses; E.H.-O., H.B., and R.A.B. performed skeletal muscle experiments; T.J.M., N.N., E.H.-O., R.A.B., M.F.S., D.L.M., H.M.C. analyzed data; T.J.M., H.M.C. wrote the paper; T.J.M., N.N., E.H.-O., J.D.L., R.A.B., M.S., D.L.M., H.M.C. edited the paper; T.J.M., D.L.M., H.M.C., M.F.S. obtained funding.
Corresponding author
Ethics declarations
Competing interests
T.J.M. and H.M.C. have filed a patent application through Columbia University based on this work. T.J.M. and H.M.C., 2019. Composition and methods for genetically-encoded high voltage-activated calcium channel blockers using engineered ubiquitin ligases (U.S. Application Serial No. 62/830,142). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Morgenstern, T.J., Nirwan, N., Hernández-Ochoa, E.O. et al. Selective posttranslational inhibition of CaVβ1-associated voltage-dependent calcium channels with a functionalized nanobody. Nat Commun 13, 7556 (2022). https://doi.org/10.1038/s41467-022-35025-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35025-7
This article is cited by
-
Aberrant expression of NEDD4L disrupts mitochondrial homeostasis by downregulating CaMKKβ in diabetic kidney disease
Journal of Translational Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
