
Abstract
Electrochemical nitrate reduction to ammonia offers an attractive solution to environmental sustainability and clean energy production but suffers from the sluggish *NO hydrogenation with the spin–state transitions. Herein, we report that the manipulation of oxygen vacancies can contrive spin−polarized Fe1−Ti pairs on monolithic titanium electrode that exhibits an attractive NH3 yield rate of 272,000 μg h−1 mgFe−1 and a high NH3 Faradic efficiency of 95.2% at −0.4 V vs. RHE, far superior to the counterpart with spin−depressed Fe1−Ti pairs (51000 μg h–1 mgFe–1) and the mostly reported electrocatalysts. The unpaired spin electrons of Fe and Ti atoms can effectively interact with the key intermediates, facilitating the *NO hydrogenation. Coupling a flow−through electrolyzer with a membrane-based NH3 recovery unit, the simultaneous nitrate reduction and NH3 recovery was realized. This work offers a pioneering strategy for manipulating spin polarization of electrocatalysts within pair sites for nitrate wastewater treatment.
Similar content being viewed by others

Introduction
Ammonia (NH3) as the critical feedstocks for artificial fertilizers production and a carbon−free energy carrier is of great significance to the modern society1,2. Nowadays, global NH3 demand exceeds 150 million tons per year, which heavily relies on the energy−intensive Haber−Bosch process associated with the consumption of 1%–2% global energy, concurrently contributing to 1.4% of global carbon emissions3,4. Recently, electrochemical reduction of nitrogen (N2) to NH3, inspired by the natural microbial N2 fixation, has attracted tremendous interest5,6,7. However, the robust N≡N (941 kJ/mol) and poor solubility in water medium of N2 lead to a low NH3 yield rate (less than 10 mmol gcat−1 h−1) and partial current densities (less than 1 mA cm−2)8,9, hindering its widespread commercialization. Alternatively, nitrate (NO3−) can be dissociated into deoxygenated species with a much lower energy of 204 kJ/mol, making it as a more suitable nitrogen source for NH3 electrosynthesis10,11, especially regarding that NO3− is widely distributed in industrial wastewaters and polluted groundwater, resulting in the eutrophication and the disturbance of ecosystems12,13. Therefore, selective reduction of NO3− to NH3 (NITRR) offers a promising route to synchronously relieve energy and environmental crisis.
Generally, NITRR initiates from the adsorption of nitrate (*NO3) on electrodes followed by the sequential deoxygenation of NO3– to adsorbed nitrite (*NO2) and nitric oxide (*NO)14,15. Subsequently, *NO hydrogenates to the hydrogenated *NHO/*NOH species with a spin state transition16,17,18. Finally, the hydrogenated species can stepwise hydrogenate to the hydroxylamine (*NH2OH) and the targeted product of *NH314,15,19. It has been reported that both the adsorption of NO3– and the subsequent hydrogenation of *NO are crucial for the rapid NITRR10,20,21,22,23,24, and thus the enhancement of NO3– adsorption and/or the acceleration of *NO hydrogenation would be effective to improve the activity of NITRR. Currently, the enhanced NO3– adsorption has been widely reported by the surface modification10, heteroatom doping20, and alloying21 of electrodes. While the acceleration of spin–transition related *NO hydrogenation via properly manipulation the spin states of electrocatalysts is seldom investigated, although this process is similar with reported oxygen−related electrocatalytic reactions such as oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) processes25,26, and generally demands tremendous energy and is kinetically sluggish, thus strongly retarding the transformation of NO3–−to−NH3. Therefore, it is urgent to design advanced spin–polarized electrocatalysts to boost the spin−dependent electron transfer for superior NITRR.
Spin−polarization of metal active sites has been reported to be an effective way to accelerate the spin−state transition between reactant intermediates, because spin−polarized metal active sites are able to drive quantum spin exchange interaction and offer a spin electron transfer channel towards spin−dependent electrocatalytic reaction27,28,29. In light of this principle, paramagnetic iron (Fe) has been extensively investigated, especially for oxygen−related electrocatalytic reactions (e.g., OER and ORR), owing to its well-tunable spin degree of electronic freedom29,30,31,32. For instance, low spin (t2g5 eg0) and intermediate spin states (t2g4 eg1) of Fe are more favorable to penetrate the antibonding orbital of O2 as compared with the high spin state of Fe (t2g3 eg2), thus facilitating the spin transition from the single−state OH−/H2O to triplet−state O231,32. More impressively, Fe single atoms on carbon matrix were found to be effective for NITRR9, exhibiting decent nitrate electroreduction capacity with an NH3 Faradaic efficiency of ~75% and a yield rate of ~20000 μg h−1 mgcat−1. Obviously, finely manipulating the spin polarization effect of Fe–based materials can provide a solution to superior NITRR.
In this study, Fe single atoms were anchored on the inherent surface oxide layer of titanium foam, where the oxygen vacancies (OVs) of titanium oxide could trigger the synchronous spin polarization of Fe and the adjacent Ti atoms. The designed spin−polarized Fe1−Ti pairs delivered a high NH3 Faradic efficiency of 95.2% and an impressive NH3 yield rate of 272000 μg h−1 mgFe−1 at −0.4 V vs. RHE for NITRR, far superior to the counterpart monolithic electrode with spin−depressed Fe1−Ti pairs (51000 μg h–1 mgFe–1) and the mostly reported NITRR electrocatalysts. The spin electrons located in the 3d orbitals of Fe and Ti atoms in the spin–polarized Fe–Ti pair sites were found to dominantly contribute this excellent performance via interacting with the key intermediates, boosting the deoxygenation of NO3– and the hydrogenation of *NO. The developed monolithic electrode with spin−polarized Fe1−Ti pairs was also employed to construct a NITRR flow−through electrolyzer coupled with a membrane separation unit for a high−efficient nitrate conversion at an industrial−level current intensity and in−situ high−purity ammonia recovery from nitrate−containing wastewater.
Results
Manipulating spin polarization of Fe1 − Ti pairs
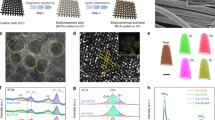
Fe single atoms anchored on a monolithic Ti electrode with different spin polarization degrees were prepared by regulating the OVs of the inherent surface oxide layer of titanium foam, as illustrated in Fig. 1a, b. The core of the synthesis process lies in the control of the reductive/oxidative gaseous environment during the thermal treatment (Supplementary Fig. 1, See details in Methods). Specifically, a reductive atmosphere (H2/Ar) was performed to create oxygen vacancies (OVs) in the inherent oxide layer of Ti electrode to evoke the spin polarization of Fe single atoms and adjacent Ti atoms (Supplementary Fig. 2), thus producing spin−polarized Fe1−Ti pairs (SP−Fe1−Ti), while an oxidative atmosphere (O2/Air) was employed to avoid the generation of OVs to obtain the spin−depressed Fe1−Ti pairs (SD−Fe1−Ti) for comparison. During hydrogen treatment of inherent oxide layer of Ti foam, electrons were first transferred from hydrogen (H) atoms to the oxygen (O) atoms in the lattice of inherent oxide layer. Then, the lattice O leaves with the H atom to form H2O, as evidenced by an obvious H2O evolution peak at about 370 °C during the temperature programmed reaction (TPR) measurement of pristine Ti foam in 5% H2/Ar (Supplementary Fig. 3), and the OVs form on the surface of TiOx/Ti foam. Electron spin resonance (ESR) spectra was utilized to identify the electronic states trapped in OVs (Fig. 1c), which clearly showed the successful creation of OVs in the SP−Fe1−Ti electrode, and the absence of OVs in SD−Fe1−Ti was also confirmed by its silent ESR signals33. As evidenced by high−resolution transmission electron microscopy (HRTEM) images of SP−Fe1−Ti and SD−Fe1−Ti (Supplementary Figs. 4, 5), the distinct boundaries were observed between the interior crystalline Ti and surface amorphous titanium oxide layer (TiOx), consistent with our previous work34. The aberration−corrected high−angle annular darkfield scanning transmission electron microscopy (HAADF−STEM) image demonstrated that Fe single atoms (marked by white circles) were well dispersed in the oxide layer of Ti foam for both SP−Fe1−Ti and SD−Fe1−Ti (Fig. 1d and Supplementary Fig. 6), and the Fe clusters or nanoparticles were not observed, consistent with the absence of metallic Fe diffraction peaks in the X−ray diffraction (XRD) pattern (Supplementary Fig. 7). The 3D surface intensity profile along the yellow arrow further confirmed the atomically dispersed Fe on the TiOx/Ti foam (Supplementary Fig. 8). The elemental EDS mapping images revealed the uniform dispersion of Fe atoms in SP−Fe1−Ti and SD−Fe1−Ti (Fig. 1e and Supplementary Fig. 9).

Schematic illustration of the manipulation strategy of spin polarization for Fe1–Ti pairs on inherent oxide surface of Ti foam for a spin–polarized Fe1−Ti (SP−Fe1−Ti) and b spin–depressed Fe1−Ti (SD−Fe1−Ti). Orange, yellow, cyan, blue, grey and transparent balls represent spin–polarized Fe, spin–depressed Fe, spin–polarized Ti, spin–depressed Ti, oxygen and oxygen vacancy (OV), respectively. c ESR spectra of TiOx/Ti, SD−Fe1−Ti and SP−Fe1−Ti. d HAADF−STEM image of SP−Fe1−Ti, where Fe single atoms were indicated by a white circle. e HADDF image and STEM elemental mapping of SP−Fe1−Ti. f FT−EXAFS spectra of SP−Fe1−Ti, SD−Fe1−Ti, Fe foil and Fe2O3 at the Fe K−edge. k3−weighted WT−EXAFS spectra of g SP−Fe1−Ti and h SD−Fe1−Ti at the Fe K−edge.
We further utilized X−ray absorption fine structure spectrometry (XAFS) to confirm the atomic dispersion of Fe atoms on TiOx/Ti. As evidenced in Fig. 1f, the Fourier−transformed Fe K−edge extended X−ray adsorption fine structure (FT−EXAFS) spectroscopy of SP−Fe1−Ti manifested a dominant peak at 1.5 Å, corresponding to Fe−O coordination in the first shell35. The absence of Fe−Fe scattering path at 2.2 Å ruled out the metallic Fe aggregation in SP−Fe1−Ti35. We also employed the wavelet−transform (WT)−EXAFS analysis to offer resolution in both radial distance and k−space and supplement the FT−EXAFS analysis. As shown in Fig. 1g, the WT−EXAFS spectra of SP−Fe1−Ti exhibited the peak with a maximum intensity at 7.2 Å−1 in contrast to Fe foil (Supplementary Fig. 10), which was assigned to the Fe−O scattering path. Similar locally fine structure of Fe atoms on the SD−Fe1−Ti electrode was also observed (Fig. 1f–h). Moreover, the electrical conductivity of SP−Fe1−Ti was very close to that of SD−Fe1−Ti (Supplementary Table 1), benefiting from the excellent electrical conductivity of Ti substrate.
Experimental evidences for the spin polarization of Fe1−Ti pairs induced by OVs
The unpaired electrons located in OVs are expected to transfer to the empty 3d levels belonging to Ti and Fe atoms adjacent to OVs36,37, resulting in two possible consequences including a negative shift in the core level binding energies of the reduced Ti or Fe atoms, and the presence of an unpaired electron (spin) in the 3d shell of the Ti or Fe atoms38,39. While the electronic properties of Fe and Ti in SD−Fe1−Ti electrode without OVs should be different from those of SP−Fe1−Ti electrode, maintaining spin−depressed Fe3+ and Ti4+ states. To confirm the electronic structures and spin states of SP−Fe1−Ti and SD−Fe1−Ti, ESR, X−ray photoelectron spectroscopy (XPS), soft X−ray absorption spectroscopy (XAS), Mössbauer spectroscopy, and temperature-dependent magnetizations (M−T) measurements were carried out. We first investigated the energy alignment of the Fermi levels (EF, see the calculation details in Supplementary Fig. 11) for TiOx/Ti, SP−Fe1−Ti, and Fe (Fig. 2a). It was found that the calculated EF of TiOx (~−3.64) was above that of Fe (~−4.5)40, so the trapped electrons at the OVs on the amorphous TiOx surface can transfer to the supported Fe atoms, resulting in the EF upshift of SP−Fe1−Ti41. As seen from the high−resolution Fe 2p core level spectra in Fig. 2b, two peaks at 710.7 and 723.8 eV were observed for SD−Fe1−Ti, which could be assigned to the Fe 2p3/2 and Fe 2p1/2 orbitals of trivalent Fe3+ species42,43. Compared with the Fe 2p spectra of SD−Fe1−Ti, the Fe 2p3/2 and Fe 2p1/2 peaks of SP−Fe1−Ti showed an obviously lower–energy shift in binding energy, suggesting the reduction of Fe valence state induced by OVs and the appearance of electron−rich Fe2+ species. As such, OVs on the amorphous TiOx surface of SP−Fe1−Ti can transfer electron to the Fe single atoms and lead to an increased electron density of Fe single atoms. Such an electron transfer from OVs to Fe atoms even occurs within FeCl3/TiOx without thermal treatment, as evidenced by the appearance of Fe2+ species in its Fe 2p XPS spectra (Supplementary Fig. 12). We further applied the Mössbauer spectroscopy to reveal the spin state of Fe single atoms on the Ti monolithic electrode (Fig. 2c), and found no sextets or singlets but doublets were derived from the deconvolution of the Mössbauer spectra for SP−Fe1−Ti and SD−Fe1−Ti, indicating the absence of Fe–Fe bonds44,45. As expected, SP−Fe1−Ti possessed a much higher content of Fe2+ species (40.2%) with high spin polarization configuration (t2g4 eg2) than SD−Fe1−Ti (26.0%) according to the fitted results (Supplementary Table 2).

a Energy alignment of the Ef for TiOx/Ti, SP−Fe1−Ti, and Fe foil. b Fe 2p XPS spectra of SD−Fe1−Ti and SP−Fe1 − Ti. c Mössbauer spectra for SP−Fe1−Ti and SD−Fe1−Ti. d Ti L−edge soft−XAS spectra of SD−Fe1−Ti and SP−Fe1−Ti. e Ti 2p XPS spectra of SD−Fe1−Ti and SP−Fe1−Ti. f 1/χm versus temperature plots and the calculated μeff of TiOx/Ti, SD−Fe1−Ti and SP−Fe1−Ti.
Furthermore, we found that OVs on both TiOx/Ti and SP−Fe1−Ti were accompanied by the appearance of spin−polarized Ti3+ species with an EPR signal at g = 1.989, which was invisible in the EPR spectra of SD−Fe1−Ti without OVs (Fig. 1c). This difference indicated that the introduction of OVs could trigger the spin−polarization of Ti sites. We thus employed the Ti L−edge soft−XAS measurement that is highly sensitive to charge state, orbitals occupation and the spin orientation of electrons46,47,48 to reveal the formation of spin−polarized Ti3+ species on SP−Fe1−Ti (Fig. 2d). The multiple spectral features of SD−Fe1−Ti were very similar to that of SrTiO3 indicating Ti4+ valence state49. In comparison with SD−Fe1−Ti, the lower−energy shift and higher L2 peak intensity of SP−Fe1−Ti verified the formation of spin−polarized Ti3+ species induced by OVs47, which was further confirmed by the shift to lower binding energy in Ti 2p XPS spectra for both TiOx/Ti and SP−Fe1−Ti (Fig. 2e).
We therefore carried out the temperature−dependent magnetic susceptibility (M−T) measurement to unravel the total effective magnetic moment (μeff), which can be determined via the Curie–Weiss law50,51. According to the 1/χm versus temperature plots of TiOx/Ti, SP−Fe1−Ti and SD−Fe1−Ti (Fig. 2f), the order of calculated μeff was in the sequence of SP−Fe1 − Ti (μeff = 6.57) > SD−Fe1−Ti (μeff = 3.46) > TiOx/Ti (μeff = 1.20), suggesting that SP−Fe1−Ti possessed much more spin electrons than SD−Fe1−Ti and TiOx/Ti. Taken together, these above results evidently showed that the spin polarization of atomically dispersed Fe1−Ti pairs can be easily manipulated by OVs, providing an opportunity for developing unique spin−polarized Fe−based single atom catalysts towards NITRR and deepening insights into spin effect on NITRR.
Evaluation of NITRR performance towards different electrodes
NITRR performance was investigated on different electrodes in a H−type electrolytic cell under ambient conditions. For the linear sweep voltammetry (LSV) in 1 M KOH (Fig. 3a), the addition of nitrate much more increased the current density of SP−Fe1−Ti electrode than those of Ti foam, TiOx/Ti and SD−Fe1−Ti electrodes. We further quantified the NH3 Faradaic efficiency (FENH3) and NH3 yield rate at a certain potential after 2 h of electrolysis via ultraviolet-visible (UV–Vis) spectrophotometry (Supplementary Figs. 13 and 14). As expected, the SP−Fe1−Ti electrode exhibited higher FENH3 and NH3 yield rate than the SD−Fe1−Ti electrode (Fig. 3b). Impressively, the SP−Fe1−Ti electrode displayed an attractive NH3 yield rate of 272000 μg h−1 mgFe−1 (16 molNH3 gFe–1 h–1) and a high Faradaic efficiency of 95.2% at −0.4 V vs. RHE, which were also verified by the results obtained from 1H nuclear magnetic resonance (NMR, Supplementary Fig. 15) measurement. Such an excellent NITRR activity of SP−Fe1−Ti electrode was much superior to those of state−of−the−art NITRR electrocatalysts, Haber−Bosch catalysts and N2 reduction electrocatalysts (Fig. 3c and Supplementary Table 3). Additionally, SP−Fe1−Ti electrode possesses promising application potential for maximizing FENH3, onset potential and current density at certain potential (Supplementary Table 4) over almost all of top−level NITRR electrodes consisting of the low−price components (e.g., Cu, Ti, and Fe in Supplementary Table 5). We also drop−casted SP−Fe1−Ti catalyst powders on the carbon fiber paper with the catalyst mass loading of 1 mg cm−2 for performance assessment. As shown in Supplementary Fig. 16, SP−Fe1−Ti catalyst displays a high cathodic current density of 190 mA cm−2 at −0.4 V vs. RHE, superior to most of the top−level NITRR catalysts. The NH3 yield rate defined by the electrode area of SP−Fe1−Ti catalyst was determined to be 0.99 mmol cm−2 h−1, which outperforms most of low−price metal−based catalysts and even several high−price metal−based catalysts (Supplementary Table 6). More importantly, the monolithic nature of SP−Fe1 − Ti electrode is much more feasible than the powder form of previous top−level NITRR electrocatalysts for practical application.

a LSV curves of Ti foam, TiOx/Ti, SD−Fe1 − Ti and SP−Fe1 − Ti electrodes. b NH3 yield rate and FENH3 of SD−Fe1−Ti and SP−Fe1−Ti electrodes at various potentials. c Comparison of the electrocatalytic NITRR performance of SP−Fe1−Ti electrode with other extensively reported electrocatalysts. d NMR spectrum of the products generated during the electrocatalytic NITRR on the SP−Fe1−Ti monolithic electrode at −0.4 V vs. RHE. e Partial NH3 current densities of SD−Fe1−Ti and SP−Fe1−Ti electrodes normalized to the geometric area at various potentials. f Specific activity normalized to ECSA of TiOx/Ti, SD−Fe1 − Ti and SP−Fe1−Ti electrodes at −0.4 V vs. RHE. g NH3 yield rate and Faradaic efficiency of SP−Fe1−Ti electrode under the applied potential of −0.4 V vs. RHE during 20 consecutive electrolysis cycles.
The contributions of other possible products to the faradaic current were also evaluated using gas chromatography equipped with thermal conductivity detector (TCD) and UV–Vis spectrometry. Only H2 and NO2− were detected (Supplementary Fig. 17 and Supplementary Fig. 18), and their FEs were calculated to be 0.7% and 4.1%, respectively, at –0.4 V vs. RHE. After taking the two side−reactions into consideration, the total FE (Supplementary Fig. 19) was 100% as expected. Moreover, the NH3 selectivity of SP−Fe1−Ti electrode was determined to be 95.9% at –0.4 V vs. RHE, further confirming its high selectivity towards NH3 production from NITRR.
We thus employed NMR to confirm the N source of generated NH3 via the isotope labeling experiments with 15N−labeled NO3− as the reagent. Different from 14NH4+ with three peaks in the 1H NMR spectra, only two peaks of 15NH4+ appeared in 1H NMR spectra when 15NO3− was used as the reagent (Fig. 3d), demonstrating that the produced NH3 was from nitrate feedstock instead of contaminations. In addition, peaks of 15NH4+ are absent in 1H NMR spectra of the original electrolyte containing 15NO3−, further confirming that the produced NH3 was originated from the reduction of nitrate feedstock instead of contaminations. Remarkably, the SP−Fe1 − Ti electrode also showed a larger NH3 partial current density of 174 mA cm−2 at −0.7 V vs. RHE than the SD−Fe1−Ti electrode (Fig. 3e), suggesting a great potential in practical applications. We also assessed the intrinsic activity of SP−Fe1−Ti electrode through calculating the specific activity (SA) by normalizing the electrode activity to the electrochemical surface area52 (ECSA, Supplementary Fig. 20), and found that the SA of SP−Fe1−Ti electrode was apparently higher than SD−Fe1 − Ti and TiOx/Ti electrodes at −0.4 V vs. RHE (Fig. 3f), revealing the intrinsically high catalytic NITRR activity of spin−polarized Fe1−Ti pairs and the great contribution of spin−polarization of active metal sites to NITRR.
Since the nitrate concentration varies in different sources, we also checked the NITRR activity of SP−Fe1−Ti electrode in the electrolyte with different NO3− concentrations (0.1, 0.5, and 1 M) at −0.4 V vs. RHE. As shown in Supplementary Fig. 21, the FEs of NO3–−to−NH3 conversion were 95.1%, 96.6% and 95.2% in the electrolyte with 0.1, 0.5, and 1 M NO3−, respectively. These results suggest that the NO3– concentration has no obvious impacts on FENH3 of SP−Fe1−Ti electrode, indicating the wide NO3− concentration compatibility. In addition, we observed that the NH3 yield rate was enhanced by increasing the NO3– concentrations from 0.1 to 1 M because of the accelerated mass transfer23. We further evaluated the NITRR activity of the SP−Fe1−Ti electrode at the neutral condition. Impressively, the SP−Fe1−Ti exhibited comparable NH3 yield rate (15 mol h−1 gFe−1) and FE (96.1%) to that of at the alkaline condition (Supplementary Fig. 22), further demonstrating its high potential of environmental application.
More importantly, the FENH3 and NH3 yield rate of SP−Fe1−Ti electrode kept very stable during 20 cycles of consecutive electrolysis in a H−type electrolytic cell at −0.4 V vs. RHE (Fig. 3g and Supplementary Fig. 23), indicating its long−term durability and high potential for practical application. XRD, XPS, and TEM analyses further revealed the high stability of SP−Fe1−Ti electrode during NITRR (Supplementary Figs. 24–26), as confirmed by the negligible Fe dissolution of SP−Fe1−Ti electrode within 20 cycles consecutive electrolysis (Supplementary Fig. 27).
Insights into NITRR mechanism towards SP−Fe1−Ti pairs
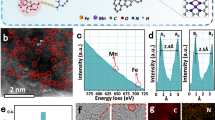
Density functional theory (DFT) calculations were employed to get insights into NITRR mechanism over SP−Fe1−Ti pairs. We first explored the density of states (DOSs) of the key NO and NHO species involved in NITRR (Fig. 4a), and confirmed the asymmetric spin state of NO in DOS of spin−up and spin−down states, which was different from that of NHO, indicating that the transformation of NO to NHO was accompanied by a spin–state transition. As revealed by the constructed models of SP−Fe1−Ti and SD−Fe1−Ti (Fig. 4b), their Fe atoms were respectively coordinated with three and four O atoms for SP−Fe1−Ti and SD−Fe1−Ti, consistent well with the fitting EXAFS results (Supplementary Fig. 28 and Table 7). The spin density diagrams displayed that only Fe atom was spin–polarized for SD−Fe1−Ti, while both Fe atom and Ti atom adjacent to Fe atom were spin–polarized in the case of SP−Fe1−Ti (Fig. 4b). This magnetic difference further verified the increase of the M−T measured magnetic moment with the existence of OV (Fig. 2f). Magnetic states were also evident from the DOSs of Fe and Ti atoms. The spin up and spin down components of Fe atom around Fermi level (EF) were quite different (Fig. 4c), indicative of unpaired electrons in the Fe−3d orbitals. Besides, the spin−up and spin−down components of Ti atom in SD−Fe1−Ti were roughly the same, but unpaired electronic states appeared near EF after the introduction of OV (Fig. 4c). These magnetic Ti and Fe atoms contributed to the highly efficient NITRR.

a DOSs of NO and NHO species. b Optimized structures and calculated 2D spin density diagrams of SD−Fe1−Ti and SP−Fe1−Ti. c DOSs of Fe and Ti atoms on SD–Fe1–Ti and SP–Fe1–Ti. d DEMS measurements of NITRR over SP–Fe1–Ti electrode. e Calculated free energy diagrams for NITRR on SD−Fe1−Ti and SP−Fe1−Ti. f Major orbital interactions between Fe atom and NO molecule as well as the corresponding molecular orbital diagrams (BD and BD* represent bonding and antibonding orbital, respectively). g Charge density differences of NO and NHO adsorbed on SD–Fe1–Ti and SP–Fe1–Ti (red and cyan represent electron accumulation and depletion, respectively). h COHPs of NHO adsorbed on SD–Fe1–Ti and SP–Fe1–Ti. i DOSs of Ti atom on SP−Fe1−Ti before and after NHO adsorption. Ti, O, Fe, N, and H atoms are denoted by grey, red, cyan, blue, and white, respectively.
Given that the hydrogen source for NITRR under alkaline condition comes from H2O, water dissociation including the HO−H bond cleavage and the proton transfer ability would strongly influence the catalytic activity of NITRR. It was found that H2O dissociation was slightly endothermic by 0.08 and 0.03 eV, with barriers of 0.28 and 0.29 eV on SD−Fe1−Ti and SP−Fe1−Ti, respectively, suggesting that H2O dissociation on the both electrodes were much favorable and comparable (Supplementary Fig. 29). We further employed the electron spin resonance (ESR) technique using 5,5−dimethyl−1−pyrroline−N−oxide (DMPO) as the radical trapping reagent to investigate the generation and the role of hydrogen radicals from water dissociation to understand the mechanism of nitrate reduction. Nine ESR peaks with an intensity ratio of 1:1:2:1:2:1:2:1:1 were observed in Supplementary Fig. 30 for the electrocatalysis on the SP−Fe1−Ti electrode in pure 1 mol L−1 KOH, which could be assigned to the spin adduct of DMPO−H, confirming the generation of Hads. When adding NaNO3 in 1 mol L−1 KOH, the signal intensity of DMPO−H disappeared. Such a phenomenon revealed that the generated Hads through water dissociation was consumed, suggesting the deep participation of H* in the hydrogeneration process of NITRR. We further conducted kinetic isotope effect (KIE) experiments to estimate the proton transfer ability of SP−Fe1−Ti and SD−Fe1−Ti during NITRR by comparing the current density ratios. The NITRR KIE value (1.40) of SP−Fe1−Ti was close to that (1.53) of SD−Fe1−Ti at −0.4 V vs. RHE (Supplementary Fig. 31), suggesting a similar hydrogen transfer ability on the two electrodes. Therefore, the water dissociation and hydrogen transfer abilities have tiny impact on the distinct NITRR activities of SP−Fe1−Ti and SD−Fe1−Ti.
To understand the unique role of spin−polarized Fe1–Ti pairs on NITRR, the NITRR reaction pathways on SD−Fe1−Ti and SP−Fe1−Ti were investigated. To construct a comprehensive description of the reaction mechanism, online differential electrochemical mass spectrometry (DEMS) was utilized to on-line detect the key intermediates and products (Fig. 4d). We observed the m/z signals of 46, 30, 31, 33, and 17 from NO2, NO, NHO, NH2OH, and NH3, respectively. On the basis of DEMS results, we calculated the free energy of individual intermediate on SD−Fe1−Ti and SP−Fe1−Ti (Fig. 4e, Supplementary Fig. 32) under a potential of 0 V vs. RHE53,54,55. Actually, NO3− to *NO underwent an NO3− adsorption step and two deoxygenation steps, namely, *+NO3− → *NO3 → *NO2 → *NO. In the case of the SD−Fe1−Ti system, the NO3− adsorption underwent an uphill Gibbs free energy change of 0.34 eV. Afterward, *NO3 first deoxygenated to *NO2 with a downhill free energy change of −1.52 eV. Subsequently, *NO2 continued to remove one oxygen to form *NO with an energy release of 1.81 eV. As for the SP−Fe1−Ti system, the above three steps release the energy of 0.64, 1.65, and 0.93 eV, respectively. Finally, NO3− deoxygenated to *NO with a total downhill Gibbs free energy change of −3.00 eV on SD−Fe1−Ti, while the total downhill free energy change for the above steps was −3.22 eV on SP−Fe1−Ti. Therefore, NO3− deoxygenation to *NO on SP−Fe1−Ti was more favorable thermodynamically. For NITRR on SD−Fe1−Ti, the potential−determining step (PDS) was *NO to *NHO with an thermodynamic barrier of 0.75 eV, while the PDS was determined to be *NH2 to NH3 with a thermodynamic barrier of 0.46 eV on SP−Fe1−Ti, consequently resulting in a higher NITRR activity than SD−Fe1 −Ti53,55,56. This PDS change stemmed from the effect of both spin–polarized Fe and Ti sites on SP−Fe1−Ti, resulting in the remarkably decreased free energy of *NO to *NHO from 0.98 eV to −0.23 eV. Meanwhile, a much smaller peak intensity ratio (~3.92) of *NO to *NHO for SP−Fe1−Ti than that of SD−Fe1−Ti (~5.83, Supplementary Fig. 33) was observed, further confirming an accelerated *NO hydrogenation process on the SP−Fe1−Ti electrode. To further highlight the synergistic effect of Fe1−Ti spin pairs, we calculated the free energy of *NHO intermediate adsorbed on the Fe1−Ti pairs involving a Ti atom without spin–polarization for comparison (Supplementary Fig. 34), and the calculated value of 0.37 eV was much higher than that on SP−Fe1 − Ti, suggesting that the spin–polarization of Ti atom was also significantly contributed the higher activity of NITRR. Subsequently, we investigated the competitive hydrogen evolution reaction on spin−polarized Fe1 − Ti pairs (Supplementary Fig. 35), and the calculated free energy for *H adsorption on Fe and Ti sites of SP−Fe1−Ti were 1.13 and 0.78 eV, respectively, much weaker than that of NO3− adsorbed on spin−polarized Fe1−Ti pairs (−0.64 eV). Therefore, the H2 generation on the spin−polarized Fe1−Ti pairs were inhibited, facilitating to enhance the Faradaic efficiency for NITRR.
To further understand the spin−polarization effect of Fe1−Ti pairs on promoting the activity of NITRR, the orbital interactions between *NO/*NHO and the Fe–Ti pair were analyzed in detail, since *NO hydrogenation was generally suggested to govern the activity24. For NO adsorption on SP−Fe1−Ti, it was found that NO preferably adsorbed on Fe (−1.80 eV), stronger than that on Ti (−1.09 eV). The main contribution to the interaction between Fe and *NO was from the dxz/dyz/dz2 orbitals of Fe atom and the σ/π* orbitals of NO (Fig. 4f), according to the high spin polarization configuration (t2g4 eg2) of Fe, as mentioned above. We noted that the dxy and dx2−y2 orbitals of Fe could not hybridize with the NO orbital owing to symmetry conservation, their orbital interaction was thus not considered57,58. Thus, in such a bonding model, the semioccupied dz2 of Fe can accept electrons from the occupied σ orbital of NO, and in turn the electrons of Fe dxz/dyz orbitals can inject into the partially occupied π* orbitals to weaken the bond order of NO57,59,60. This “donation−backdonation” of electrons generally led to the NO’s acceptance of 0.25 |e| from the substrate (Fig. 4g and Supplementary Fig. 36), resulting in a binding energy of −1.80 eV for NO adsorption on the Fe site of SP−Fe1−Ti, stronger than that of −1.58 eV on the Fe site of SD−Fe1−Ti that transferred less electron of 0.17 |e| to *NO. Moreover, the spin polarized pairs can also be advantageous of the hydrogenation of *NO through significantly stabilizing hydrogenated *NHO intermediates. The calculated adsorption energy of −2.41 eV for *NHO that preferably adsorbed on SP−Fe1−Ti via the formation of Fe–N and Ti–O was much stronger than that of −1.22 eV on SD−Fe1−Ti via forming Fe−N and Fe−O bonds, because more electrons transferred to *NHO from SP−Fe1−Ti (0.53 |e|) than that from SD−Fe1−Ti (0.39 |e|), as evidenced by the Bader charge analysis (Fig. 4g), which was supported by the Crystal Orbital Hamiltonian Populations (COHP) analysis (Fig. 4h). The negatively integrated COHP (−COHP) values for Fe−N (2.74 eV) and Ti−O (3.04 eV) interactions on SP−Fe1−Ti were more positive than those for Fe−N (2.25 eV) and Fe−O (2.70 eV) interactions on SD−Fe1−Ti, suggesting that the spin−polarized Fe1−Ti pairs exhibited a stronger interaction with *NHO. As stated before, Ti atom adjacent to Fe atom of SP−Fe1−Ti underwent a spin state transition from unspin polarization to spin polarization after the introduction of OVs, as supported by the appearance of unpaired dyz, dxy, and dz2 states near the EF (Fig. 5i). Furthermore, the dz2 spin state near the EF of Ti atom on SP–Fe1–Ti disappeared after the adsorption of *NHO, implying that the unpaired spin electron of Ti atom injected into *NHO61,62. Similarly, the unpaired spin electron in dz2 state of Fe can also partially inject into *NHO (Supplementary Fig. 37), enhancing the electron transfer from Fe–Ti spin pair to further stabilize *NHO. Conclusively, both spin−polarized Fe−Ti pair sites can effectively facilitate the deoxygenation of NO3− to *NO and stabilize *NO and *NHO intermediates to boost the subsequent *NO hydrogenation, as well as inhibit the hydrogen evolution reaction, leading to high activity and selectivity of SP−Fe1−Ti electrode towards NITRR.

a Schematic illustration of integrated equipment composed of flow−cell reactor and membrane separator by employing the SP−Fe1−Ti monolithic cathode for NITRR and a gas−permeable hydrophobic membrane for NH3 recovery. b Long−term stability test of SP−Fe1−Ti electrode at −0.4 V vs. RHE using the integrated equipment. c NH3 production and recovery efficiency of the integrated equipment during long−term stability test. d XRD pattern of the obtained NH4Cl powder.
An integrated equipment for flow−through NITRR electrolyzer and in−situ ammonia recovery
Besides the electrode performance, NH3 product separation and recovery are also crucial for the practical application of NITRR11,63,64. From this aspect, we designed an integrated device composed of a flow−through NITRR electrolyzer and a membrane−based ammonia recovery unit to realize the efficient NITRR and in−situ NH3 recovery, as schematically illustrated in Fig. 5a and Supplementary Fig. 38. We designed a compact flow−through electrolyzer consisting of SP−Fe1−Ti cathode and the mesh-type commercial dimension stable anode (DSA, the Ru0.8Ir0.2O2/Ti electrodes with a coating thickness of ~10 μm) as the NITRR electrolyzer. During operation, the NO3−−containing solution (250 mL) as synthetic effluent was treated through electrochemical reduction on SP−Fe1−Ti cathode, and circulated by a peristaltic pump with a flow rate of 60 mL min–1. Then the NH3−containing effluent was directly guided into the hollow polypropylene (PP) fiber arrays immersed into 1 M HCl solution (800 mL) to recover NH3. The key of simultaneous NITRR and NH3 product recovery is the continuous flow of NH3 through the gas−permeable hydrophobic membrane and then back to the cathode chamber. During this process, the produced NH3 could permeate through membrane between the cathode chamber and the recovery chamber and be absorbed by acid solution in the recovery chamber63,64. By using the SP−Fe1−Ti monolithic cathode, the coupled device could robustly work at an industrial−level current density of ~200 mA cm−2 for up to 200 h with a high FENH3 of 95% (Fig. 5b). As a result, this sustainable and decentralized system achieved high NITRR and simultaneous NH3 recovery with nearly 100% selectivity and about 90% recovery efficiency (Fig. 5c). After long−term electrolysis, the measured contact angle of membrane (Supplementary Fig. 39) was determined to be 123.5°, similar with that of pristine membrane (123.5°), suggesting that the post−reacted membrane still kept its hydrophobicity. After further rotary evaporation, the recycled ammonia in solution could be converted into high−purity NH4Cl powder11, as confirmed by the XRD pattern (Fig. 5d). These findings demonstrated the feasibility of selective conversion of nitrate−containing wastewater into upgraded ammonia fertilizers through combining the high−performance SP−Fe1−Ti monolithic cathode with the integrated device, providing a sustainable way for nitrogen cycle.
Discussion
In conclusion, the spin−polarized Fe1−Ti pairs were designed by manipulating oxygen vacancies of the inherent surface oxide layer on monolithic Ti electrode, and the resultant electrode could deliver extraordinary NITRR activity with an impressive NH3 yield rate (272,000 μg h−1 mgFe−1) and a high NH3 Faradic efficiency of 95.2% at −0.4 V vs. RHE, suppressing the counterpart of spin−depressed Fe1−Ti electrode and most of the well−known electrodes. It was demonstrated that spin−polarized Fe−Ti pairs could provide spin electrons to interact with the reaction intermediates, and consequently facilitate to the deoxygenation of NO3− to *NO and the hydrogenation of *NO via strengthening their adsorption. By coupling the NITRR flow−through electrolyzer with the membrane separation technology, we successfully realized the on−site ammonia recovery in the fertilizer’s formation from the nitrate−containing electrolyte in a sustainable and decentralized manner, by taking advantage of the resultant spin−polarized Fe1−Ti pairs electrode. Our study not only showcases the construction of spin−polarized Fe1−Ti pairs for NITRR and potential wastewater treatment or ammonia production applications, but also offers an innovative strategy to develop the advanced electrodes with spin polarization effect towards spin−related reactions.
Methods
Chemicals
The titanium monolithic electrode was purchased from Kunshan Guangjiayuan New Material Co., Ltd., China. Iron chloride (FeCl3·6H2O), potassium sodium tartrate (KNaC4H6O6), DMSO–d6, maleic acid (C4H4O4), sodium nitrate (NaNO3), potassium hydroxide (KOH), potassium nitrate (K15NO3) and ethanol were purchased from Sinopharm Chemical Regent Company. All chemicals were used without further purification.
Electrode preparation
The synthesis route of SP–Fe1–Ti electrode was illustrated in Supplementary Fig. 1a, as previously reported1. Typically, the TiOx/Ti monolithic electrode with 2 cm × 2 cm in size was first pretreated in reductive atmosphere (5 % H2/Ar) at 400 °C for 3 h with a heating rate of 5 °C min–1 to create the oxygen vacancies (OVs) on the electrode surface. Then, the homogeneous Fe precursor solution with a Fe mass concentration of 4 mg mL–1 in ethanol was uniformly deposited onto the as–prepared TiOx/Ti monolithic electrode. Meanwhile, the deposition process was under the infrared lamp illuminating to fasten the solvent evaporation and avoid the solvent aggregation arising from surface tension. Finally, the SP–Fe1–Ti electrode was obtained by treating the Fe–deposited electrode in reductive atmosphere (5 % H2/Ar) at 400 °C for 3 h with a heating rate of 5 °C min–1 once more. The SD–Fe1–Ti electrode followed the similar synthesis route but in an oxidative atmosphere (O2/Air) to avoid the generation of OVs for comparison (Supplementary Fig. 1b).
Characterization
X–ray diffraction (XRD) patterns were recorded by employing a Rigaku Miniflex–600 (Cu Kα, λ = 0.15406 nm, 40 kV and 15 mA). The high–angle annular dark–field scanning transmission electron microscope (HAADF–STEM) images were obtained on a JEOL JEM–ARM200F TEM/STEM with a spherical aberration corrector working at 200 kV. X–ray photoelectron spectroscopy (XPS) was carried out on scanning X–ray microprobe (PHI 5000 Verasa, ULAC–PHI, Inc.) with Al Ka radiation. The C 1 s peak at 284.6 eV is as internal standard for energy calibration. The energy dispersive X–ray spectroscopy (EDS) was performed on FEI Talos F200X. Electron spin resonance (ESR) was conducted on the EMX micro–6/1 under 100 K. The X–ray absorption fine structure spectra of Fe K–edge was obtained at Singapore Synchrotron Light Source center (SSLS, operating at 2.5 GeV with a maximum current of 200 mA). The EXAFS spectra were determined via subtracting the post–edge background from the overall absorption and then normalized to the edge–jump step. Subsequently, the χ(k) data was Fourier transformed to real (R) space by using a hanning windows (dk = 1.0 Å–1) to separate the EXAFS contributions from different coordination shells. To obtain the quantitative structural parameters around central atoms, least–squares curve parameter fitting was performed with using the ARTEMIS module of IFEFFIT software packages. XAS spectra of Ti–L edge were collected at the BL 11 A beamline of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. We measured electrical conductivities of SP−Fe1−Ti and SD−Fe1−Ti electrode at room temperature through the four probe DC technique.
Electrochemical measurements
NITRR test was conducted in a typical H–type cell, separated by a Nafion 117 membrane. The Fe1–Ti electrodes with specific spin polarization degree (SP–Fe1–Ti and SD–Fe1–Ti), platinum plate and Hg/HgO electrode were used as working electrode, counter electrode and reference electrode, respectively. If not specified otherwise, all potentials reported in this work were referenced to a reversible hydrogen electrode (RHE). Before each measurement, the electrolyte was purged with Ar (99.99%) for 30 min. The linear sweep voltammetry (LSV) curves were recorded at a scan rate of 10 mV s−1 under stirring with rotation rate of 500 rpm. Potentiostatic tests were conducted at certain applied potentials within NITRR window for 2 h. Nessler’s reagent method was employed to quantify the generated NH3 in electrolyte to further determine the faradaic efficiency (FENH3) and NH3 yield rate. In particular, a certain amount of electrolyte was taken out from the cell and diluted to the detection range. Then, an aqueous solution of potassium sodium tartrate (KNaC4H6O6, 100 μL, 500 g L–1) and Nessler’s reagent (100 μL) were added into the diluted electrolyte (5 mL) to obtain the uniform mixture. Let stand for 10 min. The UV–Vis absorption intensity at the absorbance peak at 420 nm was collected to determine the NH3 concentration of a series of standard ammonium chloride solutions and the mixed solution. The concentration–absorbance curve was plotted for the calculation of the NH3 concentration of the mixed solution (Supplementary Fig. 14). The NH3 yield rate was calculated via the following relation:
The FENH3 was calculated via the following relation:
where cNH3 is the measured NH3 concentration, V is the volume of the electrolyte, t is the reaction time, mcat is the Fe mass on the monolithic electrode, F is the Faraday constant (96485 C mol−1) and Q is the total charge. The NH3 amount was further determined by 1H NMR (600 MHz, Bruker Avance III) with 10 vol% DMSO–d6 as the spin–lock field and 0.4 mg L–1 maleic acid (C4H4O4) as the internal standard, respectively. Before each 1H NMR measurement, the pH of obtained solution was adjusted to 2.0 with HCl. The concentration of NH4+ was plotted versus the peak area ratio of NH4+ and C4H4O4 to obtain the standard curve. The generated NH4+ concentration can be determined from standard curve. An isotope–labeling experiment was performed in the electrolyte containing 1 mol L–1 KOH and 1 mol L–1 K15NO3/ K14NO3 for 2 h at −0.4 V vs. RHE to specify the nitrogen source of NH3. The obtained 15NH4+/14NH4+ was observed in 1H NMR spectra. The possible gas products were detected by a gas chromatography (GC, Thermal Trace−1300) equipped with thermal conductivity detector (TCD). The Griess test was used to determine the NO2− concentration in electrolytes after the potentiostatic test. Generally, N−1−naphthyl ethylenediamine dihydrochloride (0.04 g), p−aminobenzene sulfonamide (0.8 g), and H3PO4 (2 mL, 85%) were dissolved in Dl water (10 mL) using the Griess reagent. The diluted electrolyte (5 mL) was mixed with the Griess agent (100 uL) and rested for 10 min at room temperature to conduct UV−vis tests. NO2− concentrations of the electrolyte were determined by the absorbance at 540 nm. In the same operation, various NaNO2 aqueous solutions were used as the standard samples to obtain the calibration curve (Supplementary Fig. 17). The FE of byproduct i (FEi) was calculated via the following relation:
The selectivity of NH3 was calculated via the following relation:
where ni is the number of electrons transferred to byproduct i, ci is the measured byproduct i concentration, V is the volume of the electrolyte or the upper space, F is the Faraday constant (96485 C mol−1) and Q is the total charge.
The electrical double layer capacitor (Cdl) was determined from double–layer charging curves using cyclic voltammograms (CVs) within non–faradaic potential region between 0.4 and 0.5 V vs. RHE at different scan rates from 20 mV s–1 to 100 mV s–1. Half of the current density difference (ΔJ/2) at the centered potential was plotted against the scan rate (v) to calculate the slope of fitted straight line and obtained the Cdl value. The electrochemical active surface area (ECSA) was calculated by dividing the double–layer capacitance (Cdl) by specific capacitance (Cs, 40 µF cm–2). Differential Electrochemical Mass Spectrometry (DEMS) measurements were carried out on a specially–made electrochemical cell with electrolyte constantly flowing in through a peristaltic pump. During the test, Ar was kept bubbling into the electrolyte. The mass signals were constantly collected during the LSV measurements from 0.1 to –0.6 V at a scan rate of 10 mV s–1. When the mass signal returned to baseline, another three cycles were conducted under identical condition to avoid the accidental error.
Data availability
All data that support the findings of this study are present in the paper and the Supplementary Information. Further information can be acquired from the corresponding authors upon reasonable request.
References
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum–catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536–540 (2019).
Service, R. F. New recipe produces ammonia from air, water, and sunlight. Science 345, 610–610 (2014).
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290–296 (2019).
Tang, C. & Qiao, S. –Z. How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully. Chem. Soc. Rev. 48, 3166–3180 (2019).
Hao, Y. –C. et al. Promoting nitrogen electroreduction to ammonia with bismuth nanocrystals and potassium cations in water. Nat. Catal. 2, 448–456 (2019).
Geng, Z. et al. Achieving a record–high yield rate of 120.9 for N2 electrochemical reduction over Ru single–atom catalysts. Adv. Mater. 30, 1803498 (2018).
Lv, C. et al. An amorphous noble–metal–free electrocatalyst that enables nitrogen fixation under ambient conditions. Angew. Chem. Int. Ed. 57, 6073–6076 (2018).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Wu, Z. –Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Chen, G. –F. et al. Electrochemical reduction of nitrate to ammonia via direct eight–electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Chen, F. –Y. et al. Efficient conversion of low–concentration nitrate sources into ammonia on a Ru–dispersed Cu nanowire electrocatalyst. Nat. Nanotech. 17, 759–767 (2022).
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Duca, M. & Koper, M. T. M. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726–9742 (2012).
Wang, Y. et al. Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges. Chem. Soc. Rev. 50, 6720–6733 (2021).
Xu, H. et al. Electrocatalytic reduction of nitrate–a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710–2758 (2022).
Shafirovich, V. & Lymar, S. V. Nitroxyl and its anion in aqueous solutions: spin states, protic equilibria, and reactivities toward oxygen and nitric oxide. Proc. Nat. Acad. Sci. 99, 7340–7345 (2002).
Ford, P. C. & Miranda, K. M. The solution chemistry of nitric oxide and other reactive nitrogen species. Nitric Oxide 103, 31–46 (2020).
Janaway, G. A. & Brauman, J. I. Direct observation of spin forbidden proton–transfer reactions: 3NO– + HA → 1HNO + A. J. Phys. Chem. A 104, 1795–1798 (2000).
Gao, Q. et al. Breaking adsorption–energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine–learned insights. Nat. Commun. 13, 2338 (2022).
Zhang, S. et al. High–ammonia selective metal–organic framework–derived Co–doped Fe/Fe2O3 catalysts for electrochemical nitrate reduction. Proc. Nat. Acad. Sci. 119, e2115504119 (2022).
Wang, Y. et al. Enhanced nitrate–to–ammonia sctivity on copper–nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Cheng, X. –F. et al. Coordination symmetry breaking of single–atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper–supported rhodium cluster and single–atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Clayborne, A., Chun, H. –J., Rankin, R. B. & Greeley, J. Elucidation of pathways for NO electroreduction on Pt(111) from first principles. Angew. Chem. Int. Ed. 54, 8255–8258 (2015).
Mtangi, W. et al. Control of electrons’ spin eliminates hydrogen peroxide formation during water splitting. J. Am. Chem. Soc. 139, 2794–2798 (2017).
Do, V. –H. & Lee, J. –M. Orbital occupancy and spin polarization: from mechanistic study to rational design of transition metal–based electrocatalysts toward energy applications. ACS Nano 16, 17847–17890 (2022).
Chen, R. R. et al. Antiferromagnetic inverse spinel oxide LiCoVO4 with spin–polarized channels for water oxidation. Adv. Mater. 32, 1907976 (2020).
Liang, Y. et al. Enhancement of electrocatalytic oxygen evolution by chiral molecular functionalization of hybrid 2D electrodes. Nat. Commun. 13, 3356 (2022).
Ren, X. et al. Spin–polarized oxygen evolution reaction under magnetic field. Nat. Commun. 12, 2608 (2021).
Wu, T. et al. Spin pinning effect to reconstructed oxyhydroxide layer on ferromagnetic oxides for enhanced water oxidation. Nat. Commun. 12, 3634 (2021).
Li, X. et al. Identification of the electronic and structural dynamics of catalytic centers in single–Fe–atom material. Chem 6, 3440–3454 (2020).
Li, Z. et al. Spin engineering of single–site metal catalysts. Innovation 3, 100268 (2022).
Dai, J. et al. Single–phase perovskite oxide with super–exchange induced atomic–scale synergistic active centers enables ultrafast hydrogen evolution. Nat. Commun. 11, 5657 (2020).
Yao, Y. et al. Single atom Ru monolithic electrode for efficient chlorine evolution and nitrate reduction. Angew. Chem. Int. Ed. 61, e202208215 (2022).
Zhang, S. et al. Electrocatalytically active Fe–(O–C2)4 single–atom sites for efficient reduction of nitrogen to ammonia. Angew. Chem. Int. Ed. 59, 13423–13429 (2020).
Muller, D. A. et al. Atomic-scale imaging of nanoengineered oxygen vacancy profiles in SrTiO3. Nature 430, 657–661 (2004).
Chakraborty, T., Meneghini, C., Aquilanti, G. & Ray, S. Microscopic distribution of metal dopants and anion vacancies in Fe-doped BaTiO3−δ single crystals. J. Phys. Condens. Matter 25, 236002 (2013).
Sala, G. et al. Vacancy defects and monopole dynamics in oxygen-deficient pyrochlores. Nat. Mater. 13, 488–493 (2014).
Yang, T.-Y. et al. A new hematite photoanode doping strategy for solar water splitting: oxygen vacancy generation. Phys. Chem. Chem. Phys. 15, 2117–2124 (2013).
Wei, K. et al. Strained zero-valent iron for highly efficient heavy metal removal. Adv. Funct. Mater. 32, 2200498 (2022).
Mao, C. et al. Energy–confined solar thermal ammonia synthesis with K/Ru/TiO2–xHx. Appl. Catal. B Environ. 224, 612–620 (2018).
Xie, J. et al. Highly selective oxidation of methane to methanol at ambient conditions by titanium dioxide–supported iron species. Nat. Catal. 1, 889–896 (2018).
Mondal, P. et al. Behavior of As(V) with ZVI–H2O system and the reduction to As(0). J. Phys. Chem. C. 118, 21614–21621 (2014).
Liu, W. et al. Discriminating catalytically active FeNx species of atomically dispersed Fe–N–C catalyst for selective oxidation of the C–H bond. J. Am. Chem. Soc. 139, 10790–10798 (2017).
Zitolo, A. et al. Identification of catalytic sites for oxygen reduction in iron– and nitrogen–doped graphene materials. Nat. Mater. 14, 937–942 (2015).
Ohtomo, A., Muller, D. A., Grazul, J. L. & Hwang, H. Y. Artificial charge–modulationin atomic–scale perovskite titanate superlattices. Nature 419, 378–380 (2002).
Pan, L. et al. Manipulating spin polarization of titanium dioxide for efficient photocatalysis. Nat. Commun. 11, 418 (2020).
Chang, C. F. et al. c-axis dimer and its electronic breakup: the insulator-to-metal transition in Ti2O3. Phys. Rev. X 8, 021004 (2018).
Schlappa, J. et al. Resonant soft x-ray scattering from stepped surfaces of SrTiO3. J. Phys. Condens. Matter 24, 035501 (2012).
Guo, Y. et al. Engineering the electronic state of a perovskite electrocatalyst for synergistically enhanced oxygen evolution reaction. Adv. Mater. 27, 5989–5994 (2015).
Miao, J. et al. Spin–state–dependent peroxymonosulfate activation of single–atom M–N moieties via a radical–free pathway. ACS Catal. 11, 9569–9577 (2021).
Dai, J. et al. Hydrogen spillover in complex oxide multifunctional sites improves acidic hydrogen evolution electrocatalysis. Nat. Commun. 13, 1189 (2022).
Wang, Y. et al. Unveiling the activity origin of a copper–based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Gao Q., et al. Synthesis of core/shell nanocrystals with ordered intermetallic single–atom alloy layers for nitrate electroreduction to ammonia. Nat. Synth. https://doi.org/10.1038/s44160-023-00258-x (2023).
Liu, H. et al. Low–coordination Rhodium catalysts for an efficient electrochemical nitrate reduction to ammonia. ACS Catal. 13, 1513–1521 (2023).
Long, J. et al. Direct electrochemical ammonia synthesis from nitric oxide. Angew. Chem. Int. Ed. 59, 9711–9718 (2020).
Sun, Y. et al. Spin–related electron transfer and orbital interactions in oxygen electrocatalysis. Adv. Mater. 32, 2003297 (2020).
Wei, X. et al. Tuning the spin state of Fe single atoms by Pd nanoclusters enables robust oxygen reduction with dissociative pathway. Chem 9, 181–197 (2023).
Li, J. et al. 3.4% solar–to–ammonia efficiency from nitrate using Fe single atomic catalyst supported on MoS2 nanosheets. Adv. Funct. Mater. 32, 2108316 (2022).
Niu, H. et al. A feasible strategy for identifying single–atom catalysts toward electrochemical NO−to−NH3 conversion. Small 17, 2102396 (2021).
Gao, R. et al. Pt/Fe2O3 with Pt–Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614–623 (2021).
Liu, K. et al. Insights into the activity of single–atom Fe–N–C catalysts for oxygen reduction reaction. Nat. Commun. 13, 2075 (2022).
Gao, J. et al. Electrocatalytic upcycling of nitrate wastewater into an ammonia fertilizer via an electrified membrane. Environ. Sci. Technol. 56, 11602–11613 (2022).
Gao, J. et al. Electrochemically selective ammonia extraction from nitrate by coupling electron– and phase–transfer reactions at a three–phase interface. Environ. Sci. Technol. 55, 10684–10694 (2021).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1201701, 2022YFA150470, L.Z.), the National Natural Science Foundation of China (U22A20402, U21A20286, L.Z.), (22102100, Y.Y.), (22206121, J.D.), (22273068, U22A20394, X.G.), Shenzhen Science and Technology Program (JCYJ20220818095601002, L.Z.), the Natural Science Foundation of Shanghai (22ZR1431700, Y.Y.), the China Postdoctoral Science Foundation (2021M702117, 2022M722080, J.D.), the National Postdoctoral Program for Innovative Talents (BX20220197, J.D.) and the Shanghai Postdoctoral Excellence Program (2021182, J.D.), (2022381, G.Z.). The authors acknowledge the support from the Max Planck−POSTECH−Hsinchu Center for Complex Phase Materials, the Instrumental analysis center of Shanghai Jiao Tong University, Instrumental analysis center of School of Environmental science and engineering, Shiyanjia Lab and Bruker Corporation for the help in characterizations and experimental measurements, and Supercomputing Center of Wuhan University.
Author information
Authors and Affiliations
Contributions
Y. Y., X. G., and L.Z. Z. conceived the idea. J. D. and L. Z. carried out the experiments. Y. T. carried out the DFT calculations. J. D., Y. T., Y. Y., X. G., and L.Z. Z. wrote the paper. Z. H., C. C., C. K., G. Z., J. W., X. Z., Q. Z., W. H., R. W., K. W., and R. Z. helped with data analysis and manuscript polishing. All the authors discussed results and provided comments during the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wenhui He, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, J., Tong, Y., Zhao, L. et al. Spin polarized Fe1−Ti pairs for highly efficient electroreduction nitrate to ammonia. Nat Commun 15, 88 (2024). https://doi.org/10.1038/s41467-023-44469-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-44469-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
