
Abstract
Nitrate (NO3‒) pollution poses significant threats to water quality and global nitrogen cycles. Alkaline electrocatalytic NO3‒ reduction reaction (NO3RR) emerges as an attractive route for enabling NO3‒ removal and sustainable ammonia (NH3) synthesis. However, it suffers from insufficient proton (H+) supply in high pH conditions, restricting NO3‒-to-NH3 activity. Herein, we propose a halogen-mediated H+ feeding strategy to enhance the alkaline NO3RR performance. Our platform achieves near-100% NH3 Faradaic efficiency (pH = 14) with a current density of 2 A cm–2 and enables an over 99% NO3–-to-NH3 conversion efficiency. We also convert NO3‒ to high-purity NH4Cl with near-unity efficiency, suggesting a practical approach to valorizing pollutants into valuable ammonia products. Theoretical simulations and in situ experiments reveal that Cl-coordination endows a shifted d-band center of Pd atoms to construct local H+-abundant environments, through arousing dangling O-H water dissociation and fast *H desorption, for *NO intermediate hydrogenation and finally effective NO3‒-to-NH3 conversion.
Similar content being viewed by others


Introduction
Nitrate (NO3‒) is one of the most widespread water contaminants, sourced from agricultural runoff and industrial sewage discharges, that disharmonizes the global nitrogen cycle1,2,3,4. The presence of NO3‒ pollution in water bodies is a matter of significant environmental concern due to its detrimental impacts on both aquatic ecosystems and human health. Excessive NO3‒ concentrations can lead to eutrophication5, the depletion of dissolved oxygen, and the production of harmful algal blooms. Ingesting water with high NO3‒ levels can also have adverse health effects, inducing problems such as cancer6 and the commonly known blue-baby disease7.
To address the pressing challenges posed by NO3‒ pollution, tremendous efforts have been made to develop cost-effective methods for NO3‒ removal, ranging from reverse osmosis, ion exchange, electrocatalysis to electrodialysis and biological denitrification8,9. Among them, electrocatalysis is the most promising solution10,11,12, performed under mild conditions with high selectivity, versatility and environmental sustainability1,13,14,15,16. Within this scope, electrocatalytic NO3‒ reduction reaction (NO3RR), which converts NO3‒ to ammonia (NH3) with renewable electricity inputs, offers an operational avenue for restoring the disturbed nitrogen cycle and facilitating the denitrification of wastewater, as well as a sustainable alternative to the energy-intensive Haber-Bosch techniques that consume 2% of the world’s energy and release 1.4% of global carbon dioxide emissions17,18,19,20,21. Tackling NO3‒ in high pH systems is of particular interest owing to its practical applications toward industrial and agricultural wastewater22 and less formation of toxic nitrogen oxide byproducts23,24,25.
In the past five years, significant advancements have been made in the field of alkaline NO3RR to NH3 synthesis, utilizing a repertoire of catalysis, including Cu encapsulated in a porous carbon framework26, Fe-based single-atom catalysts (SACs)27, CuPd nanocubes24, strained Ru nanoclusters28, reduced-graphene-oxide-supported RuCu alloy29, and Cu50Ni50 alloy30. In spite of continuous improvements in Faradaic efficiency (FE), a close-to-100% value was only reported under moderate NH3 yield rates (<10 mg h−1 cm−2) and limited current density (<300 mA cm−2)28,29,30,31. Further increasing the current density and production rate have been accompanied by a degradation of the FE18,23. Given the pressing challenge of wastewater containing NO3‒ resulting from urbanization and population growth, alkaline electrocatalytic system for NO3‒ reduction with both high NH3 FE (>95%) and fast reaction rates (>1 A cm−2) is of pivotal importance for a sustainable future, but remains elusive.
In this study, we design and realize a halogen-mediated alkaline electrocatalytic platform to overcome the limitation and achieve high-speed conversion of NO3‒ to NH3 while maintaining an ideal FE. Modifying Pd species on Cu2O platform with excellent NO3‒ adsorption and conversion ability32,33 could favor H2O dissociation under high pH conditions34,35,36. But the intense interaction between the d orbitals of Pd and s orbitals of *H (* denotes the adsorbed state) brings strong Pd-H binding37, which affects the desorption of *H. Halogen elements with high first-electron-affinity38 can tailor the 3d orbital electron structure of Pd atom to regulate *H release, thereby breaking the bottleneck of FE due to the scarce proton (H+) feeding in high pH conditions. Here we develop Cl-coordinated Pd SACs-dispersed Cu2O matrix (Pd-Cl/Cu2O) nanocrystal to carry out alkaline NO3RR. As a result, we simultaneously achieve a NH3 FE of ~100% with a current density of ~2 A cm−2 for 1 h and NH3 yield rate of ~330 mg h−1 cm−2 at 1 M NO3– concentration (pH = 14), outperforming previous results with large current densities23,39,40. Impressively, our platform can reduce the NO3‒ concentration from an industrial wastewater level of 56 mM to a drinkable water level (<0.8 mM), with an over 99.1% NO3–-to-NH3 conversion efficiency. Further, we demonstrate a successful conversion of NO3– into practical ammonia products with near-unity efficiency via coupling the NO3RR with an air stripping process. The combined results of in situ Raman spectroscopy, in situ attenuated total reflection infrared spectroscopy (ATR-IR), kinetic isotope effect (KIE) experiments, and theoretical simulations reveal that Cl-coordination induces a shifted d-band center of Pd atoms to construct local H+-abundant environments, through triggering the dissociation of dangling O-H water and fast *H desorption for *NO intermediate hydrogenation and finally efficient NO3‒-to-NH3 conversion (Fig. 1). This tactic can be extended to other halogen element Pd-(F, Br, I)/Cu2O for alkaline NO3RR to NH3, demonstrating the wider applicability of the halogen-mediate strategy.

Schematic diagram of Cl mediated H+ feeding to boost *NO intermediate hydrogenation and finally achieve efficient NO3‒-to-NH3 conversion in alkaline NO3RR over Pd-Cl/Cu2O.
Results
Theoretical prediction
To test the halogen mediating effect, density functional theory (DFT) calculations were conducted on the models of Cu2O, Pd-dispersed Cu2O (Pd/Cu2O) and Pd-Cl/Cu2O with different Cl numbers (Supplementary Figs. 1, 2). Cu2O showed a high Gibbs free energy change for H2O dissociation (\(\Delta {G}_{{*{{\mbox{H}}}}_{2}{{\mbox{O}}}}\) of 1.19 eV, Fig. 2a), while the corresponding value on Pd/Cu2O (0.70 eV) decreased, indicating the accelerated H2O dissociation under the assistance of Pd SACs. The introduction of Cl coordination endowed Pd-Cl/Cu2O model (optimized model of Pd atoms coordinated with two Cl, Supplementary Fig. 3) with a further decrease of \(\Delta {G}_{{*{{\mbox{H}}}}_{2}{{\mbox{O}}}}\) (0.68 eV) and more favorable to generate H+ (\(\Delta {G}_{{{{\mbox{H}}}}^{+}}\) of −1.34 eV).

a Gibbs free energy change of H+ supply over catalysts. b The charge density difference between *H intermediate and catalysts. The isosurface level was 0.005 e·bohr-3. The yellow and cyan colors represented positive and negative charge regions, respectively. 0.13 e was the electrons transferred from Pd-Cl/Cu2O to *H, and 0.09 e was the electrons transferred from Pd/Cu2O to *H. c The local density of states of Pd 3d orbital for the Pd/Cu2O and Pd-Cl/Cu2O catalysts. d Gibbs free energy diagram of various intermediates generated during NO3RR over Pd-Cl/Cu2O at the potential of −0.6 V vs. RHE for pH = 14.
To investigate the influence of Cl coordination on the H+ formation, differential charge distribution and the local density of states (LDOS) were calculated. From the charge density difference (Fig. 2b), the two Cl atoms on Pd-Cl/Cu2O both obtained 0.47 e, which was supplied by the coordinated Pd atom (0.13 e) in addition to the contribution from the Cu2O substrate. The Cl ligand with strong first-electron-affinity seized the electrons of Pd38 and induced a downward shifted 3dxy and \(3d_{{z}^{2}}\)-band center of Pd atom in Pd-Cl/Cu2O compared to that of Pd/Cu2O, as well as a upward shifted 3dxz-band center of Pd (Fig. 2c). Thus, under the regulation of Cl ligand, the shifted d-band center of Pd enabled *H on the catalyst to obtain more electrons (0.13 e) and make *H more unstable to promote the H+ release (Fig. 2b).
Next, after considering the effects of potential and pH on NO3RR pathway with multiple possible branches (Supplementary Figs. 4–6), the optimal pathway on Cu2O, Pd/Cu2O and Pd-Cl/Cu2O models at the potential of −0.6 V vs. RHE for pH = 14 was proposed and the corresponding ∆G of each intermediate was calculated (Fig. 2d and Supplementary Figs. 7, 8). In such a sequential electron–proton transfer process (Supplementary Fig. 9)41,42, the hydrogenation of *NO2 into *NO2H (*NO2 + H2O + e‒ → *NO2H + OH−) was the potential-determining step (PDS), which involved a ∆G of −0.57 eV over pure Cu2O. Pd/Cu2O also presented a relatively lower ∆G of PDS (−0.65 eV). Pd-Cl/Cu2O showed the lowest ∆G of PDS (−0.76 eV) and correspondingly advanced the progress of NO3RR. Thus, Pd-Cl/Cu2O was anticipated as a promising candidate for alkaline NO3RR towards NH3 synthesis.
Synthesis and structural characterizations of catalysts
Inspired by the theoretical results, we synthesized Cu2O, Pd/Cu2O and Pd-Cl/Cu2O catalysts (see details in Methods). Scanning electron microscopy (SEM), transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) showed Cu2O had a lotus-like morphology on Cu foam with the orientation of (111) crystal planes (Supplementary Figs. 10–15). Pd-Cl/Cu2O and Pd/Cu2O maintained similar morphology to the initial Cu2O (Fig. 3a and Supplementary Figs. 16, 17). X-ray diffraction (XRD) patterns only presented the peaks of Cu2O and no Pd signals in these catalysts (Supplementary Fig. 18). The corresponding energy-dispersive X-ray (EDX) elemental mapping indicated the uniform distribution of Pd species on the Cu2O matrix (Fig. 3b and Supplementary Fig. 19). The Pd appearing as the bright and isolated atoms was observed by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) images (Fig. 3c and Supplementary Fig. 20), confirming the successful preparation of Pd SACs in Pd-Cl/Cu2O and Pd/Cu2O.

SEM (a), EDX mapping (b), and AC-HAADF-STEM images (c) of Pd-Cl/Cu2O. Pd K-edge XANES spectra (d), and FT k2-weighted EXAFS spectra (e) of Pd-Cl/Cu2O and reference samples. f The fitting EXAFS spectra of Pd-Cl/Cu2O. Inset: fitting model.
To explore the electronic structure of Pd SACs, high-resolution X-ray photoelectron spectroscopy (XPS) measurements were performed. The Cu 2p XPS and Cu LMM Auger spectra (Supplementary Fig. 21a, b) identified that Cu1+ existed in all samples43,44. Pd 3d spectra displayed the binding energies of Pdδ+ (0 < δ < 2) species in Pd-Cl/Cu2O positively shifted compared with Pd/Cu2O (Supplementary Fig. 21c)45, indicating the electron overflow on Pd in Pd-Cl/Cu2O. In the Cl 2p spectra, obvious Cl signals of Pd-Cl/Cu2O proved the presence of Cl, while not observed in Pd/Cu2O (Supplementary Fig. 21d). The binding energies of Cl peaks negatively shifted 0.3 eV compared to those of commercial PdCl2 (Supplementary Fig. 22), revealing the electron-enriched Cl species in Pd-Cl/Cu2O46,47. Combined with the results of Cl 2p and Pd 3d spectra, it can be concluded that the electron transferred from Pd to Cl in Pd-Cl/Cu2O under the Cl mediation, consistent with DFT calculation analysis.
To ascertain the coordination structure of catalysts, X-ray absorption fine structure (XAFS) was investigated. The Pd K-edge X-ray absorption near edge structure (XANES) spectra exhibited the pre-edge absorption energy of Pd-Cl/Cu2O and Pd/Cu2O located between those of Pd foil and PdCl2 references (Fig. 3d and Supplementary Fig. 23a), implying the valence state of Pd within 0 to 2. The Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) suggested the absence of Pd-Pd scattering (2.5 Å) in both catalysts, verifying the single-atom dispersion of Pd. The main peak at 2.1 Å for the Pd-Cl/Cu2O can be deconvoluted into 1.8 Å and 2.2 Å scattering (Fig. 3e), ascribing to Pd-Cl and Pd-Cu coordination structures, respectively. The quantitative least-squares best-fitting of EXAFS spectra (Fig. 3f and Supplementary Table 1) confirmed that Pd center was coordinated with ~2 Cl atoms and ~3 Cu atoms (Fig. 3f, inset). By comparison, Pd atom in Pd/Cu2O only coordinated with ~3 Cu atoms (Supplementary Figs. 23b, c and Table 2). The high-resolution wavelet transform (WT) EXAFS plots in K spaces further demonstrate the existence of Pd-Cl coordination in Pd-Cl/Cu2O, but absence in Pd/Cu2O (Supplementary Fig. 24). These results suggested the successful synthesis of the Pd-Cl/Cu2O and Pd/Cu2O catalysts, as proposed in DFT simulation.
NO3 ‒ intermediates hydrogenation
To probe H+ feeding on halogen-mediated samples, in situ Raman spectra were carried out (Supplementary Fig. 25). For the Pd-Cl/Cu2O (Fig. 4a), the stretching vibration peak of H2O at 1615 cm−1 appeared at 0.1 V vs. RHE and then shifted to 1605 cm−1 as the potential decreased to −0.8 V vs. RHE, indicating a weaker H-bond of interfacial H2O to enable H2O dissociation48,49,50. Notably, the emerging H3O+ peak (1770 cm−1) proved that the *H generated by H2O dissociation was immediately desorbed from Pd-Cl/Cu2O surface, to construct local H+-abundant environments in high-pH conditions (Fig. 4a, b)51,52. The formed H+ could stably accumulate on the local cathode surface. To distinguish the type of dissociated H2O, we analyzed the envelope peaks at 3000-3700 cm−1 which can be deconvoluted into three types of interfacial H2O, including tetrahedrally coordinated water (tetra-H2O, 3230 cm−1), trihedrally coordinated water (tri-H2O, 3450 cm−1) and dangling O-H bonds of water (dangling O-H, 3600 cm−1), respectively36,48,50. The three types of interfacial water can vary as a function of electrode potential due to the Stark effect. At more negative potentials, the changes in peak intensity and shift of these interfacial water would accordingly become obvious. Compared to the tetra-H2O and tri-H2O, dangling O-H water exhibited a smoother area change and a steeper shift slope (−23.0 cm−1 V−1) as the potential decreases (Fig. 4c–e), certifying the preferential dissociation of dangling O-H water on Pd-Cl/Cu2O. Although rapid dissociation of dangling O-H water also occurred over Pd/Cu2O (Supplementary Figs. 26, 27), the non-detected H3O+ peaks suggested the difficulty in donating H+ (Supplementary Fig. 26a), due to the strong binding of *H on Pd without Cl mediation. In addition, the inapparent shift of H2O peak (1617 cm−1) and the negligible H3O+ peak manifested the poor H2O dissociation with little H+ coverage on Cu2O (Supplementary Fig. 28). Therefore, under the mediation of Cl, Pd-Cl/Cu2O catalysts could promote dangling O-H water dissociation to construct the local H+-abundant environments in alkaline conditions.

a In situ Raman spectra of Pd-Cl/Cu2O. b Schematic diagram of the local H+-abundant environment construction over Pd-Cl/Cu2O. In situ Raman spectra of Pd-Cl/Cu2O (c), corresponding peak area (d) and Raman shift (e) of various interfacial H2O structures. In situ ATR-IR spectra of Pd-Cl/Cu2O (f) and Pd/Cu2O (g) catalysts. Si-O signal was derived from the reduction of surface SiO2 on the Si semi-cylindrical prism substrate under the applied potentials.
To validate the effective H+ for *NO intermediates hydrogenation in alkaline NO3RR over catalysts, in situ ATR-IR was carried out (Supplementary Figs. 29–31). Under the driven of applied potential from 0.2 to −0.7 V vs. RHE, the detected N-O peaks (at 1540 cm−1)53 in the spectra of Pd-Cl/Cu2O demonstrated the deoxygenation of NO3‒ to the intermediate *NO. The conspicuous peaks of hydro-nitrogen intermediates (-NH) at 3200–3380 cm−1 indicated the effective hydrogenation of *NO intermediates on Pd-Cl/Cu2O54,55,56,57. In comparison, Pd/Cu2O displayed the stronger *NO intermediate peaks and the weaker -NH signals (Fig. 4f, g). These results evidenced that Cl-mediated H+ feeding could boost *NO intermediates hydrogenation in alkaline NO3RR for promising NH3 synthesis over Pd-Cl/Cu2O.
Alkaline NO3RR performance
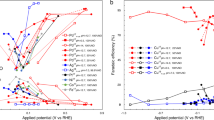
The electrocatalytic NO3RR performance was conducted under ambient temperature and pressure in a standard three-electrode H-type cell. NH4+, NO3‒, and NO2– in the reaction system were detected and quantified by colouration and 1H nuclear magnetic resonance (NMR) experiments (Supplementary Figs. 32–36). Given the common industrial and agricultural wastewater-relevant NO3‒ concentration ranging from 40 to 80 mM58,59,60,61, we reasonably selected 56 mM NO3‒ in the electrolyte (pH = 14) for the standard electrochemical tests. Linear sweep voltammetry (Supplementary Fig. 37) curves of Pd-Cl/Cu2O presented the distinct cathodic reduction peak between 0 and −0.5 V vs. RHE in the NO3‒-containing electrolyte relative to NO3‒-free solutions, expressing the underlying NO3RR process. The NO3RR performance was then determined by chronoamperometry (Supplementary Fig. 38). Pd-Cl/Cu2O delivered an excellent NH3 yield rate of 30.1 mg h−1 cm−2 with a corresponding NH3 FE of 99.2% and a current density of 350 mA cm−2 at −0.4 V vs. RHE (Fig. 5a and Supplementary Fig. 38c), in which the NH3 yield rate value was 5-folds and 2.1-folds than that of Cu2O and Pd/Cu2O. The electrochemical surface area-normalized NH3 yield also verified the best internal activity of Pd-Cl/Cu2O (Supplementary Fig. 39, 40).

a NH3 yield rate and NH3 FE of catalysts in a 1 M KOH with 56 mM NO3– electrolyte (pH = 14) for 1 h electrolysis. Catalyst mass loading: 3 mg cm−2. Resistance of catalyst: 0.156 Ω cm−2. Resistance of electrolyte: 1.45 Ω. b Kinetic isotopic effect (KIE) diagram for the ratio of NH3 yield rate in H2O to D2O solvent in a 1 M KOH with 56 mM NO3– electrolyte at −0.4 V vs. RHE. c NO3‒ removal of catalysts measured in a 1 M KOH with 56 mM NO3– electrolyte (equals 790.3 μg mL−1 NO3––N) at −0.4 V vs. RHE. After 1 h electrolysis, only 7.1 μg mL−1 of NO3––N and 0.85 μg mL−1 of NO2––N remained, both below the WHO regulations for drinking water (NO3––N < 11.3 μg mL−1 and NO2––N < 0.91 μg mL−1). d NH3 partial current densities of Pd-Cl/Cu2O in a 1 M KOH electrolyte with 1000 mM NO3– under the potential range from −0.2 to −0.6 V vs. RHE. e NH3 yield rate of Pd-Cl/Cu2O in a 1 M KOH electrolyte with different NO3– concentrations for 1 h of electrolysis. f NO3RR performance comparison of reported electrocatalysts. g Schematic of the ammonia product synthesis process from 1000 mM NO3– electrolyte to NH4Cl for 5 h electrolysis at −0.6 V vs. RHE. h The conversion efficiency of different steps for the ammonia product synthesis process. Numbers on the x-axis indicated the corresponding conversion steps in panel g. Error bars indicate the relative standard deviations of the mean (n = 3). See “Methods” for experimental details.
The KIE experiment which used D2O solvent for replacing the H2O in the electrolyte was tested to further investigate the NO3RR performance (Fig. 5b and Supplementary Fig. 41)62,63. The Cu2O presented a higher KIE value (ratio of NH3 yield rate in H2O to D2O) of 2.3, elucidating that the sluggish H2O dissociation limited the H+ offer to hinder NH3 activity. After introducing Pd SACs, the sharply decreased KIE over Pd/Cu2O (1.23) and Pd-Cl/Cu2O (1.05) corroborated the accelerated H2O dissociation, matching with the in situ Raman results. While, the lower NH3 yield rate of Pd/Cu2O than that of Pd-Cl/Cu2O resulted from the strong binding of *H on Pd, which induced the side reaction of hydrogen-hydrogen dimerization (Supplementary Figs. 41, 42). Under the Cl-mediated H+ feeding effect, Pd-Cl/Cu2O obtained superior alkaline NO3RR performance. Control experiments further demonstrated that the mediated effect originated from the Cl ligand of Pd-Cl/Cu2O rather than the free Cl ions in the system (Supplementary Figs. 43, 44).
The NO3‒ removal ability over the catalysts was examined by carrying out conversion tests under 56 mM NO3– at −0.4 V vs. RHE. The three catalysts, Pd-Cl/Cu2O, Pd/Cu2O and Cu2O, all showed a high NO3– conversion rate of ~99%, indicating that Cu2O matrix has a strong NO3– removal ability. Yet, the NO3– to NH3 conversion rates of Cu2O and Pd/Cu2O within 1 h electrolysis were as low as ~20% and ~45%, respectively, accompanied by producing 26% and 21% of NO2– (Supplementary Fig. 45). Electrochemical online differential electrochemical mass spectrometry (DEMS) showed that Cu2O and Pd/Cu2O also generated the gas products during the potentiostatic process at −0.4 V vs. RHE (Supplementary Fig. 46a, b), including the m/z signals of NH3 (17), H2 (2), N2 (28), NO (30), NH2OH (33), and N2O (44). Notably, nearly all the NO3– was converted into NH3 on Pd-Cl/Cu2O within 1 h electrolysis, and the corresponding selectivity reached ~99.1% with negligible NO2– and gas products (Fig. 5c and Supplementary Fig. 46c). The residual NO3– and NO2– concentrations were both significantly lower than the World Health Organization (WHO) regulations for drinking water (Supplementary Fig. 47)64,65. These results corroborated the excellent NO3‒ removal rate and high NH3 selectivity of Pd-Cl/Cu2O. In addition, Pd-Cl/Cu2O behaved with favorable NO3RR stability during ten-cycling tests (Supplementary Fig. 48). The morphology, phase structure, chemical valence state, atomic coordination environment and electrochemical properties remained steady after the electrolysis, further indicating the robust structure of catalysts (Supplementary Figs. 49–52 and Supplementary Tables 3, 4).
Furthermore, the optimal Pd-Cl/Cu2O (Supplementary Table 5 and Supplementary Figs. 53, 54) was utilized to explore the NO3RR performance at different NO3– concentrations, due to the diverse pollutant sources with a broad scope of NO3– concentrations1,66. Aside from 56 mM NO3–, 5, 100, 500, and 1000 mM NO3– were also chosen to cover the concentration range expected in household and heavy-industry wastewater (Supplementary Fig. 55, 56). The catalyst not only exhibited an excellent NH3 FE (~97.8%) in 5 mM NO3– system (Supplementary Fig. 57), but also preserved the high NH3 selectivity of >95% under higher NO3– concentration (100-1000 mM). Impressively, Pd-Cl/Cu2O achieved an industrial-relevant NH3 partial current density of ~2 A cm−2 (Fig. 5d) while maintaining 99.1% NH3 FE, with a splendid NH3 yield rate of ~330 mg h−1 cm−2 at 1000 mM NO3– concentration systems (Fig. 5e), which outperformed almost all state-of-the-art NO3RR performance ever reported (Fig. 5f and Supplementary Table 6)18,23,28,29,31,39,40,66,67,68.
With the impressive NO3RR performance of Pd-Cl/Cu2O catalyst, the high-purity ammonia products were collected to demonstrate their practical application potential (Fig. 5g). After conducting NO3RR tests in a sealed reactor, the cathodic electrolyte was transferred into a conical flask. The generated NH3 was striped at 70 °C by an Ar stripping method owing to the high NH3 vapor pressure in the alkaline environment (see details in Methods)69,70. As a result, ~95.2% of the NH3 vapor was successfully stripped out from the electrolyte (Fig. 5h), indicating water source denitrification with high efficiency and simultaneous production of high value-added NH3. The outflowing NH3 gas was trapped in a HCl solution (~93.1%), subsequently performed rotary evaporation, and finally collected ~80.5% of NH4Cl powder. While the high-purity NH4Cl powder (confirmed by XRD measurement in Supplementary Fig. 58) sheds light on the potential as a fertilizer for agricultural production, the limitations induced by additional cost of nitride concentration, interference (such as heavy metals and CODs) removal and product recovery17 needs to be considered and overcome for practical ammonia products.
In addition to Cl mediated strategy, this performing principle with high NH3 performance was available for other halogen ligand systems, such as Pd-F/Cu2O, Pd-Br/Cu2O and Pd-I/Cu2O. These catalysts were successfully synthesized using a similar wet-immersion and H2 calcination approach, as proved by XRD and XPS characterization (Supplementary Figs. 59–62). Besides, Pd-(F, Br, I)/Cu2O catalyst exhibited a higher NH3 activity and selectivity for alkaline NO3RR than those of Cu2O matrix and Pd/Cu2O. Their NH3 yield rates in a 1 M KOH with 56 mM NO3– electrolyte at −0.4 V vs. RHE were ~3.7, ~3.1, ~2.6-fold of Cu2O, and ~1.7, ~1.4, ~1.2-fold of Pd/Cu2O, respectively (Supplementary Fig. 63a). The corresponding NH3 FE value were ~2.9, ~2.3, ~2.1-fold of Cu2O, and ~1.3, ~1.2, ~1.1-fold of Pd/Cu2O, respectively (Supplementary Fig. 63b). Therefore, halogen-mediate strategy has expansive universality in enhancing alkaline NO3RR performance.
In summary, we have proposed and realized a highly efficient halogen-mediated H+ feeding strategy to boost the NO3RR to NH3 synthesis in alkaline conditions. Nitrate reduction to NH3, instead of N2, significantly enhances the product value of the electrochemical process. NH3 can serve as a valuable nitrogen source in agricultural fertilizers, a chemical feedstock for various industrial processes, and a high-energy-density carrier for renewable hydrogen. The optimal Pd-Cl/Cu2O nanocrystals achieved a NH3 partial current density of ~2 A cm−2 while maintaining a nearly 100% NH3 FE with a high NH3 yield rate of ~330 mg h−1 cm−2 at 1 M NO3– concentration (pH = 14). Our platform displayed an over 99.1% NO3–-to-NH3 conversion efficiency from a typical industrial wastewater level to a drinkable water level. Further, it delivered a conversion of NO3– into practical NH4Cl products with near-unity efficiency. Through a combination of theoretical simulations, in situ Raman, in situ ATR-IR, and KIE experiments, we have gained insights into the underlying mechanisms responsible for such highly selective and active NH3 synthesis over Pd-Cl/Cu2O. The presents of Cl ligand induced a prominent shift in the d-band center of Pd atoms, facilitating dangling O-H water dissociation and fast *H desorption. The constructed local H+-abundant environments supported the free H+ feeding to *NO intermediate hydrogenation, and thus realizing efficient NO3‒-to-NH3 conversion. The success of the halogen-mediated strategy presented in this study paves the way for the utilization of other halogen elements, such as F, Br, and I, in Pd-(F, Br, I)/Cu2O catalyst systems for alkaline NO3RR to NH3. The broader applicability of this approach demonstrates its potential for achieving sustainable NH3 synthesis in alkaline conditions and inspiring innovated design of environmentally friendly technologies in the field of water treatment and environmental remediation.
Methods
Chemicals
Salicylic acid (C7H6O3), trisodium citrate dihydrate (Na3C6H5O7·2H2O), sodium hydroxide (NaOH), sodium hypochlorite (NaClO), hydrochloric acid (HCl, 38%), potassium nitrate (14KNO3), potassium nitrite (KNO2), potassium iodide (KI), and ethanol (C2H6O) were purchased from Sinopharm Chemical Reagent Co., Ltd. Sulfanilamide (C6H8N2O2S), p-dimethylaminobenzaldehyde (PDAB), sodium nitroferricyanide (III) dihydrate (Na2Fe(CN)5NO·2H2O), N-(1-Naphthyl) ethylenediamine dihydrochloride (C12H14N2·2HCl), potassium hydroxide (KOH), potassium nitrate (15KNO3), dimethyl sulfoxide (DMSO-d6), maleic acid (C4H4O4), ammonium chloride (14NH4Cl, 15NH4Cl), palladium chloride (PdCl2), palladium diacetylacetonate (Pd(O2CCH3)2), palladium oxide (PdO), palladium bromide (PdBr2), palladium nitrate Pd(NO3)2, and palladium(II) trifluoroacetate (Pd(O2CCF3)2) were purchased from Aldrich Chemical Reagent Co., Ltd. All reagents were analytical reagent grades and used as received without further purification. The water used in this research was purified through a Millipore system.
Preparation of Cu2O
In a typical procedure, Cu foam (10 × 15 × 0.5 mm3) was ultrasonically washed with acetone, 2 M HCl, ultrapure water and ethanol to clean the surface, respectively. The dried pre-treated Cu foam was then anodized in a 3 M KOH solution to form the blue Cu(OH)2 nanowires by galvanostatic deposition at 20 mA cm−2 for 900 s. The brick red lotus-like Cu2O was synthesized by annealing Cu(OH)2 nanowires in a tube furnace at 550 °C for 2 h in N2 atmosphere. The Cu2O substrate plays a dual role in the catalysis. On one hand, it effectively facilitates the adsorption, activation and conversion of nitrate. On the other hand, it serves as a platform for loading Cl-coordinated Pd single atoms and provides Cu active sites to boost the NO3RR process.
Preparation of Pd-Cl/Cu2O
A certain amount of 0.03 M PdCl2 solution was immersed into the Cu foam-supported lotus-like Cu2O (10 × 10 mm2), followed by reduction at 170 °C for 15 min in 10% H2/Ar atmosphere to obtain the Pd-Cl/Cu2O catalyst. Pd-Cl/Cu2O catalysts with various Pd loadings were prepared according to the above procedure by changing the additional amount of PdCl2 solution. The Cl acted as a synthetic directing agent to stabilize Pd single atoms.
Preparation of Pd/Cu2O
A certain volume of 0.03 M Pd(O2CCH3)2 solution was soaked into the Cu foam-supported lotus-like Cu2O (10 × 10 mm2), followed by reduction at 250 °C for 15 min in 10% H2/Ar atmosphere to obtain the Pd/Cu2O catalyst.
Preparation of Pd-F/Cu2O, Pd-Br/Cu2O and Pd-I/Cu2O
For the synthesis of Pd-F/Cu2O, a certain amount of 0.03 M Pd(O2CCF3)2 was dropped into the Cu foam-supported lotus-like Cu2O (10 × 10 mm2), followed by heating at 250 °C for 15 min in 10% H2/Ar atmosphere. The Pd-Br/Cu2O or Pd-I/Cu2O were synthesized under the same procedure except that Pd(O2CCF3)2 was replaced by PdBr2 or Pd(NO3)2 and KI.
Electrochemical testing
Before the NO3RR tests, Nafion 117 membrane was pretreated as followed: first oxidizing in 5% H2O2 solution at 80 °C for 1 h to eliminate organic impurities, next boiling in deionized (DI) water for 1 h to clean the redundant H2O2 and reach the expansion, then using 0.5 M H2SO4 at 80 °C for 1 h to remove metallic impurities and residual ammonia contaminations and to protonate the membrane, finally operating DI water to rinse the excess acid and further expand the Nafion 117. The procedure should be repeated at least every 3 days to reuse the membrane.
The NO3RR was measured on an electrochemical workstation (PARSTAT 4000) with a three-electrode system in a typical H-type cell, including as-prepared catalyst electrodes (working electrode, WE), platinum electrode (counter electrode, CE), and a saturated calomel electrode (reference electrode, RE). Nafion 117 membrane was fixed between the anode and cathode cells, and each cell contained 32 mL of 1 M KOH with 56 mM NO3‒ electrolyte). All potentials reported in this work were referred to RHE scale via calibration by the following equation: \(E({{{\mbox{vs.}}}}\,{{{\mbox{RHE}}}})=E({{{\mbox{vs.}}}}\,{{{\mbox{SCE}}}})+0.244+0.0591 \times {{{\mbox{pH}}}} {{{\mbox{value}}}}\). The theoretical potential for nitrate reduction to ammonia was 0.69 V vs. RHE at pH = 1430,71. The error bars were the relative standard deviations obtained by at least three repeated tests. The CV-activation before reaction was conducted to remove impurities on the electrode surface. For the chronoamperometry measurement, the potential was applied from −0.2 to −0.6 vs. RHE. LSV was carried out in a voltage window from 0.2 to −1.0 V vs. RHE at scan rates of 10 mV·s−1.
Detection and quantification of NH3 using UV-vis
The concentration of NH3 was spectrophotometrically detected by the salicylic acid method72. In detail, the electrolyte from the cathode cell was collected and diluted to the detection range. Then, 2 mL of diluted sample was mixed with 2 mL of 1 M NaOH solution containing 5 wt% salicylic acid and 5 wt% sodium citrate. Subsequently, 1 mL of 0.05 M NaClO and 0.2 mL of C5FeN6Na2O solution (1 wt%) were added to the mixture component and shaken well. After stewing for 2 h, UV-vis spectrophotometer measurements were performed with the range from 500 to 800 nm and recorded the absorbance at the wavelength of 655 nm. The concentration-absorbance curve was calibrated using standard NH4Cl solution with concentrations of 0.1, 0.5, 1.0, 2.0, 3.0, 5.0 and 10.0 µg mL−1 in 1 M KOH with NO3‒. And the fitting curve (y = 0.104x − 0.014, R2 = 0.999) displayed a good linear relation of absorbance value with NH4+ concentration.
Detection and quantification of NO3 ‒ using UV-vis
The electrolyte from the cathode cell was collected and diluted to the detection range. 5 mL of diluted sample solution was mixed with 0.1 mL of 1 M HCl. After stewing for 20 min, the UV-vis absorbance at the wavelength ranging from 215 to 280 nm was detected73,74. The intensities at wavelengths of 220 and 275 nm were recorded, and the final absorbance difference was calculated using the equation: A = A220 nm − A275 nm. The concentration-absorbance difference curve was calibrated using standard KNO3 solution with 5, 10, 15, 20, 25, 30 and 50 µg mL−1 concentrations. And the fitting curve (y = 0.051x + 0.013, R2 = 0.999) displayed a good linear relation of absorbance value with NO3‒ concentration.
Detection and quantification of NO2 ‒ using UV-vis
The configuration of color reagent was as follows73,74: First, 0.5 g of sulfonamide was dissolved in 50 mL of 2.0 M HCl solution, which was marked as reagent A. Then, 20 mg of N-(1-naphthyl) ethylenediamine dihydrochloride was dispersed in 20 mL of DI water, which was denoted as reagent B. Subsequently, 0.1 mL of reagent A was dropped into 5 mL of standard or diluted sample solutions, mixing up and stewing for 10 min. Furthermore, 0.1 mL of reagent B was injected into the above solution, shaking up and resting for 30 min. The UV-vis absorbance at the wavelength ranging from 400 to 640 nm was recorded, in which the typical absorption peak of NO2‒ was located at 540 nm. The concentration-absorbance difference curve was calibrated using standard KNO2 solution with concentrations of 0.05, 0.1, 0.2, 0.5, 1.0, 2.0 and 3.0 µg mL−1. And the fitting curve (y = 0.768x − 0.012, R2 = 0.999) displayed a good linear relation of absorbance value with NO2‒ concentration.
Detection and quantification of NH3 using 1H NMR
To support the UV-vis results, 14NO3‒ and 15NO3‒ isotope labeling experiments were conducted on Bruker AVANCE III HD NMR spectrometer (600 MHz). The pH value of the diluted electrolyte after NO3RR was adjusted to 2 with 1 M HCl. Then 0.5 mL of the above solutions was mixed with 0.1 mL DMSO-d6 with 0.04% C4H4O4, which served as a solvent and maleic acid C4H4O4 as the internal standard. 1H NMR was recorded to quantitatively analyze of NH3 product according to the corresponding standard curves.
Electrochemical in situ Raman spectroscopy
In situ Raman was tested by inVia Reflex (Renishaw, UK) with a 633 nm laser as the excitation source. The NO3RR was performed in the custom-made Teflon reactor with a quartz window, in which the Ag/AgCl (Supplementary Fig. 25), Pt wire, and catalysts coated on Au electrode were used for the reference, counter, and working electrode, respectively. In situ Raman spectra were recorded in electrolytes with NO3‒ by the potential from open circuit potential (OCP) to −0.8 V vs. RHE.
Electrochemical in situ ATR-IR spectroscopy
ATR-IR was measured on a Nicolet iS50 FT-IR spectrometer equipped with an MCT detector and cooled by liquid nitrogen during the electrochemical process (Supplementary Fig. 29). The NO3RR was performed in the custom-made reactor with three-electrode, in which the Ag/AgCl and Pt wire were used for the reference and counter electrode, respectively. The working electrode was prepared as followed: First, the Si semi-cylindrical prism was polished with Al2O3 powder and sonicated in acetone and deionized water. The Si was pretreated in a piranha solution at 60 °C for 20 min to clean the organic contaminants. Then the reflecting surface of Si was plated in the Au precursor mixture at 60 °C for 10 min, obtaining the Au-coated Si (20 mm in diameter) conductive substrate. Finally, the catalyst ink was dropped on the substrate reflecting surface for employment in the reaction. In situ ATR-IR spectra were recorded in electrolyte with NO3‒ by the potential from 0.3 V to −0.7 V vs. RHE. The spectrum collected at OCP was used for background subtraction.
Electrochemical online DEMS tests
The online DEMS tests were performed in customized reactors containing 1 M KOH with 56 mM NO3‒ electrolyte. Ar was continuously bubbled into the electrolyte. Catalysts coated on breathable film with gold plating layer, Pt wire, and saturated Ag/AgCl electrode were used as the working, the counter and the reference electrode, respectively. After the baseline of the mass spectrometry kept steady, the potential of OCP and −0.4 V vs. RHE were applied alternately with an interval of 3 min. Accordingly, the differential mass signals appeared when the gaseous products formed on the electrode surface. The mass signal returned to baseline after the electrochemical measurement was over. To avoid accidental errors, the next cycle started using the same conditions. After five cycles, the experiment was ended.
Direct ammonia product synthesis
To evaluate the NH3 removal efficiency via Ar stripping and the NH3 collection efficiency by acid trap, 50 mL of cathodic electrolyte after NO3RR test was sealed in a conical flask at 70 °C and flowed in 100 sccm Ar gas for 5 h to perform the Ar stripping to purge the NH3 gas out. The outlet gas stream was meanwhile purged into 50 mL of 2 M HCl to collect the NH3 product. The amount of NH3 in all the solutions was measured by the salicylic acid method mentioned above, and the removal efficiency and collection efficiency were calculated as following equations, respectively:
To produce the NH4Cl product and estimate the production efficiency, the 50 mL of HCl with the trapped NH3 was dried by rotary evaporator at 70 °C in an oven overnight. The final NH4Cl was measured by a balance and analyzed by XRD. The collection efficiency of NH4Cl from the acid trap was calculated by following equation:
DFT computational details
All calculations were carried out by spin-polarized DFT with the Vienna Ab initio Simulation Package (VASP)75,76. Electron exchange-correlation was expressed by the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA)77. To describe the ionic cores, the projector augmented wave (PAW) pseudopotential was applied78,79. The Monkhorst–Pack K-points were set to be 2 × 2 × 1 for geometry optimization and density of states (DOS) calculations. The plane wave energy cutoff, and convergence criterion for electronic energy and forces were set as 450 eV, 10−5 eV, and 0.02 eV/Å, respectively (Supplementary Fig. 64). A vacuum layer of 15 Å was adopted in the models80. Aqueous phase H2O and NO3‒ were as the energetics references.
Cu2O with crystal planes (111) was modeled with a periodic 4-layer, where the lower two layers were fixed and the upper two layers were relaxed. The model included 64 Cu atoms and 32 O atoms. The optimized lattice constants were a = 12.09 Å and b = 10.47 Å, the thickness of this model was 8.79 Å. The computational hydrogen electrode (CHE) model was used to calculate the change in Gibbs free energy (ΔG)81. In CHE model, H+ + e‒ ⇌ 1/2 H2(g) was equilibrated at 0 V vs. the reversible hydrogen electrode (RHE) at all pH values.
Constant-potential calculations were applied using the code freely available from Duan and Xiao82. At the applied potential (U) on the standard hydrogen electrode (SHE) scale, the number of electrons and the atomic coordinates of the system are optimized simultaneously. The chemical potential of the electron (μ̅e) is calculated as
where μe,SHE is the electronic chemical potential of the system relative to the SHE.
where Ef is the Fermi level, Vsol is the potential deep in the solution, and φ0 = –4.6 eV for the SHE. The grand canonical energy of the system is defined as
where EDFT is the energy calculated from the DFT and Δn is the number of electrons added or removed from the system.
The chemical potential of the electron (μ̅e) is derived as
where Ω is the grand canonical energy.
Calculation of thermodynamic corrections
The zero-point energy for each species is calculate by
where vi is the vibration frequency. The entropy contributions of translational, rotational, vibrational, and electronic motion can be calculated by
where \(N\) is the number of unpaired electrons, \(R\) is the gas constant, \(P\) is the pressure, kB is the Boltzmann constant83.
From the above formula, considering that the vibration frequency of the catalyst substrate is small, its corresponding correction is very small and does not affect the calculation results. Thus, we mainly made corrections to gas molecules and adsorbents on the catalyst, and entropic effects of the catalyst substrate would not be considered further.
H2O as the proton source in alkaline conditions. The pH has an influence on the major proton donor in the research system. For alkaline nitrate reaction pathways (pH = 14), we have considered H2O as the proton source. Under this condition, the H* path will be through the alkaline pathway.
For electrochemical steps in nitrate reduction, the free energy changes are calculated using the products and reactants of the following reaction equation:
The calculation method for pH and potential effects. The free energies of adsorption of ionic species are calculated using thermodynamic Hess cycles, which cycles include the effects of entropy, solvation energy, protonation energy (including pH effects) and potential effects. The Gibbs energy formulas for nitrate reduction reaction steps are reported by Muhich et al.16,18,84. The method is based on that of Calle-Vallejo et al.85 and Liu et al.86. The free energy of anion A‒ is calculated according to:
Where \({E}_{*}\) and \({E}_{{{{\mbox{A}}}}^{*}}\) are the DFT computed enthalpies of bare surface and A* adsorbed to the surface, respectively. \({G}_{{{{\mbox{H}}}}_{2}}\) and \({G}_{{{\mbox{HA}}}}\) are the Gibbs free energies of desorbed species H2 and HA, respectively, in the gas phase at 300 K, as calculated from the following:
where EHA is the DFT computed energy of HA in the gas phase, T is the temperature (300 K), EZPE is the contribution of the zero-point energy, S is the entropic contributions to the free energy obtained using the JANAF database. The solvation energy is described:
The pH accounts for the effects on the free energies of the species. Free energy modifications due to pH were calculated according to:
GHA(g) and GHA(l) are the free energies of HA molecule in the gas and liquid phases respectively. k is the Boltzmann constant. Ka is the acid dissociation constant for the A‒ anion. The standard state (25 °C, 100 kPa, 1 mol/kg) energies of ion and neutral species in aqueous solution (GHA(g), GHA(l), \({G}_{{{{{{{\rm{A}}}}}}}^{-}}\), \({G}_{{H}^{+}}\), Ka) are taken from the CRC handbook.
The computational hydrogen electrode (CHE) is used to account for potential effects on reaction energies87:
where ΔE is the DFT computed reaction (electronic) energy, ΔEZPE and ΔS are the zero-point energy difference and the entropy difference between the adsorbed state and the gas phase, respectively. 0.0591*pH represents the free-energy contribution due to the variations in H concentration. We considered the effect of a potential bias on all states involving one electron or hole in the electrode by shifting the energy of this energy by eURHE, where URHE is the electrode potential relative to the reversible hydrogen electrode (RHE).
Data availability
All experimental data reported in this study and Supplementary Information are available from the corresponding author upon reasonable request.
References
Katsounaros, I., Dortsiou, M. & Kyriacou, G. Electrochemical reduction of nitrate and nitrite in simulated liquid nuclear wastes. J. Hazard. Mater. 171, 323–327 (2009).
Burow, K. R., Nolan, B. T., Rupert, M. G. & Dubrovsky, N. M. Nitrate in groundwater of the united states, 1991−2003. Environ. Sci. Technol. 44, 4988–4997 (2010).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Galloway, J. N. et al. Transformation of the nitrogen cycle: Recent trends, questions, and potential solutions. Science 320, 889–892 (2008).
Smolders, A. J. P., Lucassen, E. C. H. E. T., Bobbink, R., Roelofs, J. G. M. & Lamers, L. P. M. How nitrate leaching from agricultural lands provokes phosphate eutrophication in groundwater fed wetlands: The sulphur bridge. Biogeochemistry 98, 1–7 (2009).
Picetti, R. et al. Nitrate and nitrite contamination in drinking water and cancer risk: A systematic review with meta-analysis. Environ. Res. 210, 112988 (2022).
Knobeloch, L., Salna, B., Hogan, A., Postle, J. & Anderson, H. Blue babies and nitrate-contaminated well water. Environ. Health Persp. 108, 675–678 (2000).
Shrimali, M. & Singh, K. P. New methods of nitrate removal from water. Environ. Pollut. 112, 351–359 (2001).
Rezvani, F., Sarrafzadeh, M. H., Ebrahimi, S. & Oh, H. M. Nitrate removal from drinking water with a focus on biological methods: A review. Environ. Sci. Pollut. Res. 26, 1124–1141 (2019).
Meng, G. et al. NiFe layered double hydroxide nanosheet array for high-efficiency electrocatalytic reduction of nitric oxide to ammonia. Chem. Commun. 58, 8097–8100 (2022).
Zhang, S. et al. Electrocatalytic reduction of NO to NH3 in ionic liquids by P-doped TiO2 nanotubes. Front. Chem. Sci. Eng. 17, 726–734 (2023).
Zhang, W. et al. Single atomic cerium sites anchored on nitrogen-doped hollow carbon spheres for highly selective electroreduction of nitric oxide to ammonia. J. Colloid Interface Sci. 638, 650–657 (2023).
Duca, M. & Koper, M. T. M. Powering denitrification: The perspectives of electrocatalytic nitrate reduction. Energy Environ. Sci. 5, 9726 (2012).
Xu, H., Ma, Y., Chen, J., Zhang, W. X. & Yang, J. Electrocatalytic reduction of nitrate - a step towards a sustainable nitrogen cycle. Chem. Soc. Rev. 51, 2710–2758 (2022).
Qi, D. et al. High-efficiency electrocatalytic NO reduction to NH3 by nanoporous VN. Nano Res. Energy 1, e9120022 (2022).
Wu, X. et al. Contrasting capability of single atom palladium for thermocatalytic versus electrocatalytic nitrate reduction reaction. ACS Catal. 13, 6804–6812 (2023).
Van Langevelde, P. H., Katsounaros, I. & Koper, M. T. M. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Chen, F. Y. et al. Efficient conversion of low-concentration nitrate sources into ammonia on a Ru-dispersed Cu nanowire electrocatalyst. Nat. Nanotechnol. 17, 759–767 (2022).
Liao, W. et al. Boosting nitrogen activation via Ag nanoneedle arrays for efficient ammonia synthesis. ACS Nano 17, 411–420 (2023).
Liao, W. et al. Interfacial engineering promoting electrosynthesis of ammonia over Mo/phosphotungstic acid with high performance. Adv. Funct. Mater. 31, 2009151 (2021).
Ding, J. et al. Iron-doping strategy promotes electroreduction of nitrate to ammonia on MoS2 nanosheets. Inorg. Chem. Commun. 151, 110621 (2023).
Dhamole, P. B., Nair, R. R., D’Souza, S. F. & Lele, S. S. Denitrification of highly alkaline nitrate waste using adapted sludge. Appl. Biochem. Biotechnol. 151, 433–440 (2008).
Han, S. et al. Ultralow overpotential nitrate reduction to ammonia via a three-step relay mechanism. Nat. Catal. 6, 402–414 (2023).
Gao, Q. et al. Breaking adsorption-energy scaling limitations of electrocatalytic nitrate reduction on intermetallic CuPd nanocubes by machine-learned insights. Nat. Commun. 13, 2338 (2022).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. M. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultralow concentrations via an enrichment effect. Adv. Mater. 34, e2204306 (2022).
Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522–3531 (2021).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Gao, W. et al. Alloying of Cu with Ru enabling the relay catalysis for reduction of nitrate to ammonia. Adv. Mater. 35, 2202952 (2023).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Wang, Y. et al. Structurally disordered RuO2 nanosheets with rich oxygen vacancies for enhanced nitrate electroreduction to ammonia. Angew. Chem. Int. Ed. 61, e202202604 (2022).
Jiang, M. et al. Batch-scale synthesis of nanoparticle-agminated three-dimensional porous Cu@Cu2O microspheres for highly selective electrocatalysis of nitrate to ammonia. Environ. Sci. Technol. 56, 10299–10307 (2022).
Yin, D. et al. Synergistic active phases of transition metal oxide heterostructures for highly efficient ammonia electrosynthesis. Adv. Funct. Mater. 33, 2303803 (2023).
Hu, C., Zhang, L. & Gong, J. Recent progress made in the mechanism comprehension and design of electrocatalysts for alkaline water splitting. Energy Environ. Sci. 12, 2620–2645 (2019).
Valenti, G. et al. Co-axial heterostructures integrating palladium/titanium dioxide with carbon nanotubes for efficient electrocatalytic hydrogen evolution. Nat. Commun. 7, 13549 (2016).
Zhu, K. et al. Unraveling the role of interfacial water structure in electrochemical semihydrogenation of alkynes. ACS Catal. 12, 4840–4847 (2022).
Li, Y. et al. Near-surface dilution of trace Pd atoms to facilitate Pd-H bond cleavage for giant enhancement of electrocatalytic hydrogen evolution. Nano Energy 34, 306–312 (2017).
Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
Deng, X., Yang, Y., Wang, L., Fu, X. Z. & Luo, J. L. Metallic Co nanoarray catalyzes selective NH3 production from electrochemical nitrate reduction at current densities exceeding 2 A cm−2. Adv. Sci. 8, 2004523 (2021).
Fang, J.-Y. et al. Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature. Nat. Commun. 13, 7899 (2022).
He, D. et al. Regulation of the electrocatalytic nitrogen cycle based on sequential proton–electron transfer. Nat. Catal. 5, 798–806 (2022).
Koper, M. T. M. Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 4, 2710–2723 (2013).
Zhang, Y. et al. Photoelectrocatalytic reduction of CO2 to syngas via SnOx-enhanced Cu2O nanowires photocathodes. Adv. Funct. Mater. 32, 2109600 (2021).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Du, P. et al. Single-atom-driven dynamic carburization over Pd1–FeOx catalyst boosting CO2 conversion. Chem 8, 3252–3262 (2022).
Han, Y. et al. Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal. Energy Environ. Sci. 11, 2348–2352 (2018).
Zhang, B. et al. Manganese acting as a high-performance heterogeneous electrocatalyst in carbon dioxide reduction. Nat. Commun. 10, 2980 (2019).
Li, C.-Y. et al. In situ probing electrified interfacial water structures at atomically flat surfaces. Nat. Mater. 18, 697–701 (2019).
Cai, C. et al. Atomically local electric field induced interface water reorientation for alkaline hydrogen evolution reaction. Angew. Chem. Int. Ed. 62, e202300873 (2023).
Wang, Y. H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Wang, X., Xu, C., Jaroniec, M., Zheng, Y. & Qiao, S. Z. Anomalous hydrogen evolution behavior in high-pH environment induced by locally generated hydronium ions. Nat. Commun. 10, 4876 (2019).
Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
Hu, B. & Li, J. One electron makes differences:From heme {FeNO}7 to {FeNO}8. Angew. Chem. Int. Ed. 54, 10579–10582 (2015).
Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study of electrochemical nitrogen and nitrate reduction on rhodium surfaces. Angew. Chem. Int. Ed. 59, 10479–10483 (2020).
Wirz, R., Ferri, D. & Baiker, A. ATR-IR spectroscopy of pendant NH2 groups on silica involved in the knoevenagel condensation. Langmuir 22, 3698–3706 (2006).
Wiles, D. M. & Suprunchuk, T. The infrared absorption spectra of thiosemicarbazide and related compounds: NH2 and NH vibrations. Can. J. Chem. 47, 1087–1089 (1969).
Milligan, D. E. & Jacox, M. E. Matrix-isolation infrared spectrum of the free radical NH2. J. Chem. Phys. 43, 4487–4493 (1965).
Zheng, W. et al. Self-activated Ni cathode for electrocatalytic nitrate reduction to ammonia: From fundamentals to scale-up for treatment of industrial wastewater. Environ. Sci. Technol. 55, 13231–13243 (2021).
Albina, P. et al. Influence of hydrogen electron donor, alkaline pH, and high nitrate concentrations on microbial denitrification: A review. Int. J. Mol. Sci. 20, 5163 (2019).
Ahmadi, M. T., Bodaghzadeh, M., Rahimian Koloor, S. S. & Petru, M. Graphene nanoparticle-based, nitrate ion sensor characteristics. Nanomaterials Basel 11, 150 (2021).
Liu, H. et al. Electrocatalytic nitrate reduction on oxide-derived silver with tunable selectivity to nitrite and ammonia. ACS Catal. 11, 8431–8442 (2021).
Ma, W. et al. Promoting electrocatalytic CO2 reduction to formate via sulfur-boosting water activation on indium surfaces. Nat. Commun. 10, 892 (2019).
Chen, S. et al. Unveiling the proton-feeding effect in sulfur-doped Fe-N-C single-atom catalyst for enhanced CO2 electroreduction. Angew. Chem. Int. Ed. 61, e202206233 (2022).
Su, L. et al. Electrochemical nitrate reduction by using a novel Co3O4/Ti cathode. Water Res. 120, 1–11 (2017).
Chauhan, R. & Srivastava, V. C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by Ti/RuO2 anode and iron cathode. Chem. Eng. J. 386, 122065 (2020).
Wu, Z. Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Du, H. et al. Durable electrocatalytic reduction of nitrate to ammonia over defective pseudobrookite Fe2TiO5 nanofibers with abundant oxygen vacancies. Angew. Chem. Int. Ed. 62, e202215782 (2023).
Liao, P. H., Chen, A. & Lo, K. V. Removal of nitrogen from swine manure wastewaters by ammonia stripping. Bioresour. Technol. 54, 17–20 (1995).
Yuan, M.-H., Chen, Y.-H., Tsai, J.-Y. & Chang, C.-Y. Ammonia removal from ammonia-rich wastewater by air stripping using a rotating packed bed. Process Saf. Environ. Prot. 102, 777–785 (2016).
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244 (2009).
Searle, P. L. The berthelot or indophenol reaction and its use in the analytical chemistry of nitrogen. Analyst 109, 549–568 (1984).
Carvalho, A. P., Meireles, L. A. & Malcata, F. X. Rapid spectrophotometric determination of nitrates and nitrites in marine aqueous culture media. Analusis 26, 347–351 (1998).
Polatides, C. & Kyriacou, G. Electrochemical reduction of nitrate ion on various cathodes – reaction kinetics on bronze cathode. J. Appl. Electrochem. 35, 421–427 (2005).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Duan, Z. & Xiao, P. Simulation of potential-dependent activation energies in electrocatalysis: Mechanism of O–O bond formation on RuO2. J. Phys. Chem. C 125, 15243–15250 (2021).
Ochterski, J. W. Thermochemistry in Gaussian (Gaussian inc, 2000).
Gupta, S., Rivera D. J., Shaffer M., Chismar A., Muhich C. Behavior of cupric single atom alloy catalysts for electrochemical nitrate reduction: An ab initio study. ACS EST Engg. https://doi.org/10.1021/acsestengg.1023c00207 (2023).
Calle-Vallejo, F., Huang, M., Henry, J. B., Koper, M. T. M. & Bandarenka, A. S. Theoretical design and experimental implementation of Ag/Au electrodes for the electrochemical reduction of nitrate. Phys. Chem. Chem. Phys. 15, 3196 (2013).
Liu, J.-X., Richards, D., Singh, N. & Goldsmith, B. R. Activity and selectivity trends in electrocatalytic nitrate reduction on transition metals. ACS Catal. 9, 7052–7064 (2019).
Qiao, M., Liu, J., Wang, Y., Li, Y. & Chen, Z. PdSeO3 monolayer: Promising inorganic 2D photocatalyst for direct overall water splitting without using sacrificial reagents and cocatalysts. J. Am. Chem. Soc. 140, 12256–12262 (2018).
Acknowledgements
We gratefully thank the National Natural Science Foundation of China (Grant No. 22376222 for M.L., 52372253 for J.F., 22022602 for W.Liu), the Science and Technology lnnovation Program of Hunan Province (2023RC1012 for M.L.), Central South University Research Programme of Advanced Interdisciplinary Studies (Grant No. 2023QYJC012 for M.L.), and Central South University Innovation-Driven Research Programme (Grant No. 2023CXQD042 for J.F.), and China Postdoctoral Science Foundation (2023T160735 and 2022M723547 for W.Liao). We are grateful for resources from the High Performance Computing Center of Central South University. We would like to acknowledge the help from Beam Lines BL01C1 in the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) for various synchrotron-based measurements.
Author information
Authors and Affiliations
Contributions
M.L., W.Liao, J.W., C.L. and J.F. conceived the research and supervised the project. W.Liao and J.W. performed the experiments. M.L., W.Liao, J.W., S.C., Q.W., Y.C., T.L., X.W., Y.W., W.Li, T.C., C.M., H.L., Y.L., W.Liu, J.F. and B.X. analyzed the data. K.L. and G.N. carried out the DFT simulations. W.Liao and C.L. wrote the draft. All authors discussed the results and contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xijun Liu and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, W., Wang, J., Ni, G. et al. Sustainable conversion of alkaline nitrate to ammonia at activities greater than 2 A cm−2. Nat Commun 15, 1264 (2024). https://doi.org/10.1038/s41467-024-45534-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-45534-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
